Abstract
Cholestasis in children is a serious condition due to various aetiologic factors. If children with jaundice present with acholic stool, dark urine colour, or direct hyperbilirubinaemia, the patient should be evaluated urgently. Early and timely diagnosis and initiation of appropriate treatment are extremely important determinants of morbidity and mortality. In the neonatal period, idiopathic neonatal cholestasis, alpha-1 antitrypsin deficiency, cholestasis from infections, and biliary atresia are the most common causes of cholestasis. Nowadays, with the development of genetic and molecular biological studies, the diagnosis of many diseases that have previously been evaluated as ‘idiopathic‘ can be made. It is the aetiological factor that determines the prognosis. The treatment plan is created in accordance with aetiological causes and in response to symptoms such as pruritus and malabsorption: this can be surgical treatment across a diverse spectrum, from biliary diversion to liver transplantation. In this study, the aetiology, diagnosis, and treatment of cholestasis in babies and infants are reviewed in the light of current literature.
INTROUDCTION
Cholestasis is seen in every 2,500–5,000 live births. In full-term infants, jaundice that lasts >2 weeks should be evaluated; however, it should be noted that prolonged jaundice may be observed in 15–40% of healthy infants receiving breast milk.1
Cholestasis is a clinical condition in which bile production from hepatocytes or excretion through intra/extrahepatic bile ducts is impaired, resulting in bile accumulation within hepatocytes or bile ducts. Excretion of bile-excreted substances and absorption of substances that are absorbed via bile are all effected.2-5
The bile accumulating in the hepatocytes and bile ducts causes damage to these tissues. Lipids are normally degraded by pancreatic lipases with the help of bile acids, but this does not happen during cholestasis, and digestion and absorption of lipids are impaired. As a consequence of elevated serum bile acid levels, bile acid synthesis from cholesterol is depleted resulting in hypercholesterolaemia.5,6
Cholestasis can result in chronic liver dysfunction, liver transplantation, or even death. Therefore, early diagnosis, timely evaluation, and appropriate treatment are important.2,7-9
This review will especially focus on neonatal and infant cholestasis.
AETIOLOGY
In children and neonates, cholestasis can be caused by a wide spectrum of disorders. It may develop as a result of perinatal infections, congenital anomalies involving biliary tree, genetic or metabolic disorders, and multifactorial causes. 5,7,10,11 Cholestatic aetiology is summarised in Table 1.
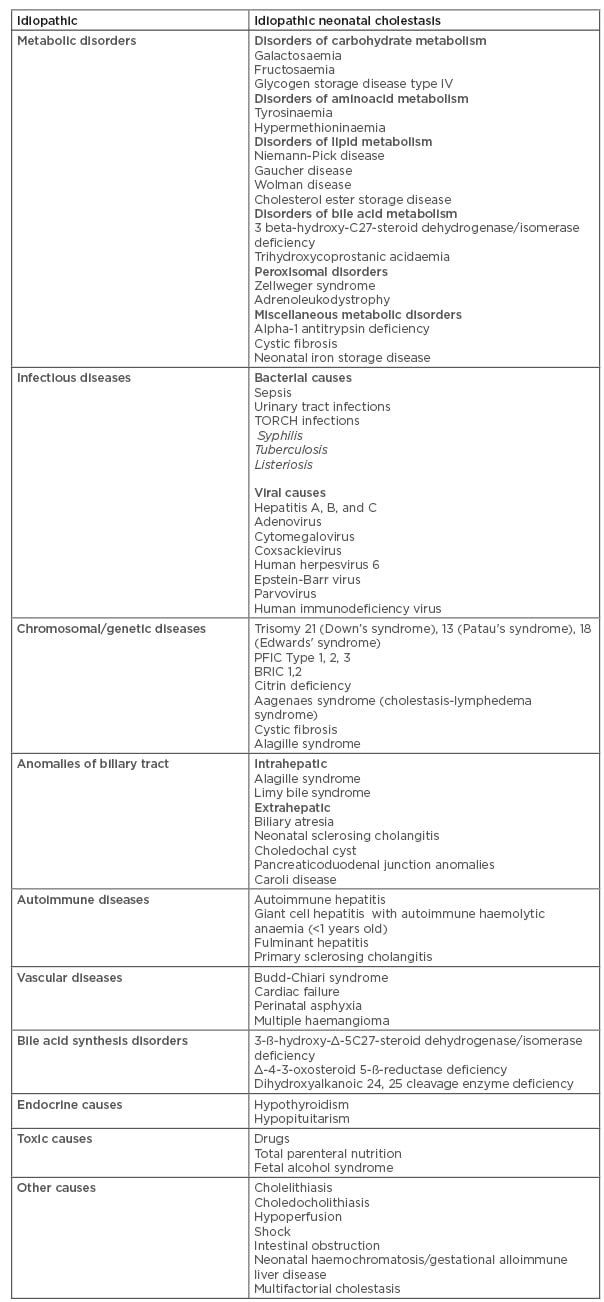
Table 1: Aetiology of cholestasis.
BRIC: benign recurrent intrahepatic cholestasis; PFIC: progressive family intrahepatic cholestasis; TORCH: toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus.
CHOLESTATIC LIVER DISEASES
The incidence of cholestatic liver disease (CLD) in newborn infants is 1 in 2,500 births. The most common causes of cholestatic jaundice in the first months of life are biliary atresia (BA), viral infections, and alpha-1 antitrypsin deficiency (A1AT).5 Cholestasis beyond the neonatal period includes a wide spectrum of congenital and acquired aetiologies.
In older children, the most common cause of biliary obstruction that leads to cholestasis is cholelithiasis. But, as mentioned above, this review will primarily focus on the differential diagnosis of cholestasis in neonates and infants. BA, choledochal cysts, tumours, and cholelithiasis result in extrahepatic cholestasis, while hepatobiliary infections; metabolic, genetic, and endocrine diseases; prematurity; drugs; parenteral nutrition; graft-versus-host disease; and post-liver transplantation pathologies can lead to intrahepatic cholestasis.
The diseases that are important causes of cholestasis are reviewed individually below.
In all chronic cholestatic disorders impaired copper secretion and accumulation can be seen.
Idiopathic Neonatal Cholestasis
Idiopathic neonatal cholestasis (INC) is the most common cause of cholestasis during the neonatal period and is responsible for approximately one third of cases. The condition appeared at a rate of 60–70% in a differential diagnosis of neonatal cholestasis in the 1980s and has now decreased to 15–20%.12 The disease is characterised by loss of appetite, jaundice, and growth retardation. The degree of cholestasis is variable and may not be differentiated from extrahepatic causes in 10% of cases. INC can be diagnosed by the presence of giant cells in liver biopsies, of which diagnostic findings are shown after 4 weeks. Patients may also have intrauterine growth retardation and prematurity. Hepatomegaly with or without splenomegaly can also be present. Before the diagnosis of INC, the integrity of the biliary tract should be evaluated and any surgical pathology should be excluded.5,12,13
Prognosis is related to the duration of cholestasis. Progressive liver disease and cirrhosis are indispensable in cholestasis lasting >6–12 months. As a result, acidity develops and the patient progresses to liver failure requiring liver transplantation.5,12
Metabolic and genetic diagnostic tools evolve, diseases once diagnosed as INC may in fact be reidentified as one of the following: primary bile acid synthesis diseases, progressive family intrahepatic cholestasis (PFIC), familial haemophagocytic lymphohistiocytosis, neonatal sclerosing cholangitis, syndromic or non-syndromic intrahepatic biliary tract paucity, neonatal haemochromatosis, or metabolic/genetic diseases such as citrine deficiency and transient neonatal cholestasis.7,9,14
Extrahepatic Billary Atresia
BA is a rare disease in which progressive inflammation and fibrosis results in cirrhosis and end-stage liver failure unless treated within the first 2 years of life. It is possible to provide bile flow with early portoenterostomy and to reduce liver damage within the first 45 days of life. Despite surgical treatment, liver injury can continue and liver transplantation may be required in 80% of patients.15,16
BA is the most common cause of liver transplantation in children; however, patients have a good 10-year survival rate after transplantation. Recent studies have focussed on therapies to prevent liver fibrosis in these patients.7,17,18
Extrahepatic BA is a cause of cholestasis in 35–41% of patients. Fibrosis and progressive obliteration of the extrahepatic biliary tract occur and lead to damage of the parenchyma and intrahepatic biliary tract. This ultimately leads to cirrhosis and death before 3 years of age. The prognosis improves with surgical management, provided that it is performed within the first 45–60 days of life.7,9,10,18,19
There are three types of atresia defined according to the alteration in the hepatobiliary system, the subtypes of which are choledochal atresia, atresia of the common hepatic duct, cystic and bile duct atresia, and atresia of the hepatic duct onwards towards the intrahepatic biliary tract, which is present in 90% of patients and is not correctable.
Usually, healthy full-term newborns begin to have jaundice and light-coloured stools between the 2nd and 6th weeks of life. They later develop firm hepatomegaly and splenomegaly. Between the 2nd and 3rd months of life, the patient’s health deteriorates with portal hypertension that progresses to cirrhosis and hepatic failure.7,17 Elevation of gamma-glutamyl transferase (GGT) is the main biochemical marker. Other laboratory findings include elevation of total bilirubin and direct bilirubin, alkaline phophatase, transaminases, and the alanine aminotransferase/aspartate aminotransferase ratio.10
Choledochal Cysts
Choledochal cysts are the congenital cystic dilatations of the biliary tree. Although they are benign, they may show malignant transformation and have serious complications such as cholangitis, pancreatitis, and cholelithiasis. Current treatment is cyst excision and hepaticojejunostomy. Despite the excision of the cyst, the potential for malignant transformation in these patients is higher than the normal population. Accordingly, long-term follow-up is necessary.10,12
Bile Acid Synthesis Disorders/Inborn Errors of Bile Acid Metabolism
More than 14 enzymes are involved in the synthesis of bile acids from cholesterol precursor molecules. Deficiencies in 3-ß-hydroxy-Δ-5C27-steroid dehydrogenase/isomerase, Δ-4-3-oxosteroid 5-ß-reductase, or dihydroxyalkanoic 24 or 25 cleavage enzymes result in the absence of synthesis of primary biliary acids resulting in cholestasis, jaundice, and early onset pruritus. Some, however, may have a more indolent presentation later during childhood.7,10,11,20 Bile acid synthetic disorders are rare forms of cholestasis, but in most cases are treatable. These conditions often present with normal or low gamma-glutamyltransferase levels. Total serum bile acids are usually low in contrast to other cholestatic disorders. Molecular techniques may identify the specific mutations in genes encoding the enzymes responsible for bile acid synthesis. Treatment with the end products of bile acid synthesis, cholic acid, and chenodeoxycholic acid, is often curative for several of the bile acid synthetic disorders.7,10,21
Metabolic Diseases
Tyrosinaemia appears as a result of cholestasis and clotting time prolongation that are not corrected after initial administration of vitamin K. It is a consequence of deficiencies of succinyl and acetone, with elevation of serum tyrosine and phenylalanine. Galactosaemia appears as malnutrition, with hypoglycaemia and reduced sugars in urine with lactose intake. It is diagnosed by measuring the levels of galactose-1-phosphate uridylyltransferase in red blood cells (without prior transfusion of red blood cells). Neonatal haemochromatosis is indicated by hepatomegaly, cholestasis, and saturation elevation of transferrin and ferritin, and is confirmed by hepatic biopsy. Wolman disease is indicated by diarrhoea, dyslipidaemia, adrenal calcifications, and cholestasis, and is diagnosed by measuring acid lipase in a skin biopsy.7,10
Alpha-1-Antitrypsin Deficiency
A1AT deficiency causes 10–34% of cholestatic jaundice in the newborn period. It occurs as the result of a mutation on chromosome 14 that leads to alteration in the production and accumulation of the A1AT protein. A1AT is a glycoprotein synthesised mainly by the liver and is the major circulating protease inhibitor that acts against neutrophil elastase. Jaundice due to autosomal recessive A1T1 deficiency usually occurs between 3 and 12 weeks. However, cholestasis is only seen in a percentage of the patients with A1T1 deficiency. The diagnosis is made by determining the serum A1AT level and is confirmed by genetic studies. There is no need to perform a liver biopsy when A1AT levels are <100 mg/dL.19,22 There is no specific treatment apart from trying to prevent the complications of chronic liver disease. Accumulation of intracellular mutant A1AT results in hepatocyte death, inflammation, fibrosis, and cirrhosis.
Infections
Some bacterial (e.g., gram-positive and gram-negative bacteraemia, urinary tract infections caused by Escherichia coli), viral (e.g., herpes simplex virus, cytomegalovirus, rubella, Epstein Barr virus), or parasitic (e.g., toxoplasmosis) infections can cause cholestasis.
Bacterial toxins have direct cholestatic action. Further release of cytokines such as IL-1 and TNF-α decrease the transport of such toxins. These molecules are fibrogenic and they directly affect the liver.
In infants, classical hepatitis viruses are not a cause of cholestasis except when there is liver failure attributable to hepatitis B virus (usually after 45 days of life).7,11,19
Genetic Diseases
PFIC is an autosomal recessive disease family with mutations in several different genes, presenting with severe pruritus and moderate jaundice commonly before 6 months of age.23-25
PFIC (Type)-1 (Byler disease) occurs from a mutation of the assumed aminophospholipid transporter gene (FIC1/ATP8B1) on chromosome 18 resulting in impaired hepatocellular bile salt secretion. PFIC-1 manifests during the infantile period with episodes of cholestasis (average onset of 3 months of age), leads to liver cirrhosis, and rapidly progresses to end-stage liver disease.
PFIC-2 occurs as a result of a mutation of the major canalicular bile salt export pump (BSEP) gene on chromosome 2 (ATP-binding cassette, sub-family B member 11 [ABCB11]).25 Expression of this gene is limited to the liver; therefore, although the clinical course of PFIC-2 is similar to that for PFIC-1, extrahepatic manifestations are absent.
PFIC-3 occurs due to a mutation of adenosine triphosphate-binding cassette subfamily B member 4 gene (ABCB4) encoding the multidrug resistance Class III (MDR3) protein related with the bile phospholipid export pump.26
GGT are low or normal in PFIC-1 and 2, whereas high in PFIC-3.
Another subtype of PFIC is defined by mutations in the nuclear receptor subfamily 1 group H member 4 (NR1H4) gene, which encodes the farnesoid X receptor (FXR), a bile acid-activated nuclear hormone receptor that regulates bile acid metabolism and balances the production and circulation of bile acids. It is presented as the master regulator of bile acid homeostasis. FXR also provides protection against hepatocarcinogenesis.23,27,28
Cholestasis is also seen in the benign recurrent intrahepatic cholestasis (BRIC) disease group, intrahepatic cholestasis of pregnancy, and erythropoietic protoporphyria.
BRIC1 may be accompanied by pancreatitis while BRIC2 may be accompanied by gallstones. The clinical features of benign recurrent intrahepatic cholestasis include early onset of recurrent attacks of cholestatic jaundice, lasting up to months in duration and eventually resolving spontaneously. It is a benign disease and fibrosis of liver cells does not occur despite repeated episodes.23
In cystic fibrosis, the liver is frequently involved and may re-present as cirrhosis, steatosis, portal hypertension, or neonatal cholestasis. However, symptoms related to cystic fibrosis-associated liver disease appear late, mainly during puberty when damage to the hepatobiliary system is already advanced.7,10
Alagille Syndrome
Alagille syndrome is a multisystem disorder related with defects in components of the Notch signalling pathway, mostly attributable to mutations in the gene JAG1. It presents with chronic cholestasis due to paucity of intrahepatic bile ducts.
Alagille syndrome is associated with cardiac alterations, butterfly vertebrae, posterior embryotoxon/pigmentary retinopathy, dysplastic kidneys, and characteristic facial deformations including hypertelorism, bulging foreheads, and prominent chins (triangular face). GGT is high in Alagille syndrome, and there is no specific treatment. Patients are at increased risk of early development of hepatocellular carcinoma, and 50% of children will require transplantation.29,30
Endocrine Disorders
Both hypothyroidism and hyperthyroidism are associated with hepatic alterations, and thyroid diseases should be excluded in patients with transaminase elevation of unknown cause. Oestrogens are related to cholestatic liver damage.31 Congenital panhypopituitarism has been recognised to cause neonatal cholestasis. Recently, isolated severe cortisol deficiency presenting with neonatal cholestasis and hypoglycaemia had been reported in which the resolution of cholestasis by hydrocortisone replacement therapy suggested a relationship between cortisol deficiency and the development of neonatal cholestasis.32
EVALUATION
History
In addition to a detailed history, diagnosis of cholestasis includes clinical findings, physical examination, laboratory studies, imaging studies, and liver biopsy. An early-as-possible diagnosis and appropriate treatment is very important; urgent complete blood count, serum direct-indirect bilirubin, blood urea nitrogen levels, electrolyte and glucose levels, prothrombin time, blood, and urine cultures should be checked.5,10,12
Clinical and Physical Findings
The most common findings of neonatal cholestasis include prolonged jaundice, acholic stool, and darkened urine. In patients with extrahepatic cholestasis, acholic stool usually appears early and pursues. If acholic stool lasts >10 days, extrahepatic aetiology should be suspected. Stool colour may show changes in intrahepatic cholestasis. Therefore, the stool colour of patients with cholestasis should be followed.33,34
Some newborns with cholestasis can present with serious problems such as intracranial haemorrhage as a result of vitamin K deficiency. Hypocalcaemia can be seen as a consequence of vitamin D deficiency and metabolic diseases (such as galactosemia, fructosemia, glycogen storage diseases), the latter of which may lead to convulsions. Panhypopituitarism can also present with cholestasis and hypoglycaemia, and lethargy and feeding problems may also be observed in metabolic disorders.5,12,34
Jaundice is the most important finding in physical examination. Hepatomegaly is usually present and may be associated with splenomegaly in advanced liver diseases.
Growth retardation as a result of intrauterine infections and facial dysmorphism as an outcome of syndromic causes may also be considered as physical examination findings.10,13,34 Choledochal cysts can show up as a mass in the right upper quadrant of the abdomen.12
Laboratory Studies
Direct bilirubin, alkaline phosphatase, and gamma-glutamyl transpeptidase are mostly elevated in CLD. Aminotransferases (aspartate aminotransferase, alanine aminotransferase) may also elevate when cholestasis persists.
If a jaundiced newborn has direct hyperbilirubinaemia, the causes of neonatal cholestasis, including BA, should be investigated. Direct hyperbilirubinaemia that is >1 mg/dL or >15% of the total bilirubin is pathological and its causes should be investigated. Indirect hyperbilirubinaemia may occur as a result of excess bilirubin production (e.g., haemolysis) or when the capacity of liver to conjugate the bilirubin is exceeded.7,10
Diagnostic Imaging Studies
Chest X-ray, abdominal ultrasonography, hepatobiliary scintigraphy, single photon emission CT, magnetic resonance cholangiography, percutaneous transhepatic cholangiography, and endoscopic retrograde cholangiography are among the diagnostic imaging methods used. The examination should start with noninvasive methods.10,35-37
Abdominal Ultrasonography
Abdominal ultrasonography, an easy and noninvasive method, is the first choice for diagnostic imaging. Ultrasonography can detect any obstructing pathologies of bile ducts, choledochal cysts, presence of gall bladder, stage of liver disease, and vascular and splenic abnormalities.
Many hepatic sonographic parameters have previously been suggested to help in the diagnosis of BA, such as the triangular cord sign (hepatic hyperechoic), abnormal gall bladder morphology, lack of gallbladder contraction after oral feeding, nonvisualisation of the common bile duct, hepatic artery diameter, hepatic artery diameter to portal vein diameter ratio, and subcapsular blood flow.14,35,36,38
Hepatobiliary Scintigraphy
Hepatobiliary scintigraphy is used to distinguish BA from other causes of cholestasis by showing the patency of the biliary tract. If 5 mg/kg/day phenobarbital is started at least 5 days prior, biliary excretion of the isotope is enhanced but has delayed diagnosis. If uptake of the labelled substances by the hepatocytes is delayed, neonatal hepatitis should be considered. Yang et al.14 compared different diagnostic methods for differentiating BA from idiopathic neonatal hepatitis (INH). In this study, liver biopsy is reported as the most reliable method to differentiate INH from BA. Among imaging methods for cholestasis, hepatobiliary scintigraphy single-photon emission computer tomography (HBS SPECT) is reported to be a more reliable diagnostic method compared to magnetic resonance cholangiography, HBS (hepatobiliary scintigraphy), or ultrasonography.14,37 Magnetic resonance cholangiography is a noninvasive technique for evaluating the intrahepatic–extrahepatic bile ducts and the pancreatic duct.39,40 Endoscopic retrograde cholangiopancreatography requires an experienced team (e.g., endoscopist, paediatric anesthesiologist) and specific infant endoscopy equipment.41
Intraoperative Cholangiography
Diagnostic laparotomy/laparoscopy and intraoperative cholangiography is the gold standard for diagnosis of BA.10 It also permits therapeutic irrigation of inspissated bile from the ductal system. If the gallbladder is visibly highly atretic, the surgeon may proceed with the Kasai procedure without the need for cholangiography. However, percutaneous transhepatic cholecysto-cholangiography can be a preferable modality for excluding BA in neonates that are critically ill or have significant comorbidities and are at a high risk during open surgery and general anesthaesia.17
Liver Biopsy
Liver biopsy is crucial in neonatal cholestasis. Bile duct proliferation, bile plugs, portal area oedema, and fibrosis at histopathological examination are typical findings of BA while bile duct paucity is seen mainly in non-BA diseases. Liver consistency is increased in BA. Within the non-BA group, lobular irregularity and prominent giant cell transformation is most prevalent in INH cases and moderate to marked interlobular bile duct injury can distinguish cases of Alagille syndrome from other non-BA cases. Nonspecific findings such as microvesicles can suggest metabolic disease.10,38,42
TREATMENT
Treatment of cholestatic patients aims to treat the underlying problem itself (either surgically or medically) and to prevent and treat the secondary complications of cholestasis. Diets, medications, and vitamins are used for medical treatment and external or internal biliary diversions are applied for surgical treatment.
Medical Treatment
Medical treatment is primarily arranged according to the aetiology. Appropriate anti-biotherapy in bacterial infections, sepsis, urinary tract infections; appropriate diet therapy in metabolic processes such as galactosaemia and tyrosinaemia; L-thyroxine treatment in congenital hypothyroidism; steroid and immunosuppressive (e.g., azathioprine, mycophenolate mofetil) treatment in autoimmune hepatitis; chelation therapy in Wilson’s disease; and effective intensive care therapy in fulminant hepatitis should be set before the patients deteriorate and have malnutrition.
The purpose of medical treatment in cholestasis can be summarised as follows: to enhance the bile flow and inhibit the accumulation of metabolites in the liver (choleresis); to treat toxic effects of bile re-entering the systemic circulation; to avoid the malabsorption of fat and fat-soluble vitamins; and to prevent acute and chronic malnutrition and ensure continuity of growth. Accordingly, the general principles of treatment regarding the secondary complications in children with CLD should include nutritional support (enough energy, protein, and medium-chain fatty acids), fat-soluble vitamins (vitamins A, D, E, and K), choleretic therapy (e.g., ursodeoxycholic acid, phenobarbital, corticosteroids), treatment of pruritus, and treatment of hypercholesterolaemia. Calcium intake and adequate exposure to sunlight are also essential.
There are many drugs used to avoid pruritus and other symptoms. Autotaxin (ATX) enzyme, a potent neuronal activator, plays a key role in the pruritogenic signalling cascade in cholestatic paediatric patients afflicted by itch in recent studies. Serum ATX activity correlated with itch intensity in children with cholestatic diseases. Lysophosphatidic acid is further formed from lysophosphatidylcholine by ATX. Serum LPA levels were found to be increased in cholestatic patients who had pruritis; however, there is no study regarding serum LPA levels in children with cholestasis.10,13, 43-46
Ursodeoxycholic acid dissolves cholestasis and is a successful treatment. Some reports suggest that, at a dose of 10–30 mg/kg per day, it could reverse the potential hepatotoxicity of the accumulating endogenous bile acids. It regulates bile acid distribution, reduces the amount of cholesterol in the bile, and provides mitochondrial integrity. It has choleretic, immunomodulatory, antioxidant, antiapoptotic, and cytoprotective effects.47-49
Cholestyramine is an oral bile acid binding resin used to resolve pruritus. It forms nonabsorbable micelles with the bile acids in the intestines and prevents bile acids from entering the enterohepatic cycle. It should be taken at least 1 hour before, or 4–6 hours after, meals, 1–4 g/day. It induces liver enzyme activity and increases bilirubin excretion. In patients with reduced serum bilirubin levels, pruritus also regresses.44
Rifampicin acts by upregulating detoxification enzymes and exporting pumps through FXR-dependent mechanisms. Ricfampicin indirectly induces hydroxylation of bile salts that are further glucuronidated and excreted in urine. It also induces conjugation and excretion of bilirubin through uridine diphosphate-glucuronosyl transferase. It is used at a dose of 5–10 mg/kg/day. 44,50
Phenobarbital is used to induce the CYP/CYP450 system in the treatment of neonatal hyperbilirubinaemia and chronic cholestasis with low bilirubin levels at a dose of 3–10 mg/kg/day.5
4-phenylbutyrate is another drug used in PFIC types. At a dosage of 350 or 500 mg/kg/day taken orally, it significantly relieves the intractable itch. In patients with decreased cell-surface expression of BSEP among PFIC-2, 4-phenylbutyrate therapy has partially restored BSEP expression at the canalicular membrane, as well as significantly improved liver tests and pruritus at a dosage of 500 mg/kg/day.15,51
Antihistaminic agents, opiate antagonists, ondansetron, steroids, propofol, and carbamazepine are included within the other medical therapy options.
Surgical Treatment
Surgery is carried out in patients whose underlying pathology causing cholestasis requires surgical manipulation, such as BA and choledochal cysts.
The most popular surgical method in BA is hepatoportoenterostomy known as the Kasai operation. This operation should be performed taking care of the micron-wide bile canaliculus at the porta hepatis. Therefore, the first 2 months are vital. Cystectomy and/or choledochoenterostomy should be performed in choledochal cysts.19,52,53
Other surgical approaches include internal or external biliary diversion surgery to relieve symptoms such as excessive pruritus. In patients who have no response to medical therapy, it is aimed to reduce the accumulation of bile by external or internal drainages. 16,53
Liver transplantation is inevitable in patients with cirrhosis and liver failure. Today, in many centres, successful donation of liver transplantation is still difficult because of religious or legal reasons.
CONCLUSION
In a large-scale systematic literature review, Catzola and Vajro54 stated that a large number of rare hepatobiliary diseases in newborns and at childhood cause cholestasis. They emphasised that, even if there is no specific or curative treatment available, immediate medical treatment and regulation of nutrition are very important to avoid complications. They also stressed that studies on inherited versus acquired cholestasis would help to develop more effective specific therapies. They emphasised that the disease-specific medical and surgical treatment approaches are life-saving.54
In conclusion, cholestasis is a fatal condition which may result in cirrhosis, liver failure, and liver transplantation. The authors believe that the most important factor affecting morbidity and mortality is the aetiology of cholestasis. Early diagnosis, timely evaluation, and appropriate treatment are important for prognosis.
As a result of the advancement in genetic and molecular biology studies, patients formerly diagnosed with INH and neonatal cholestasis have turned out to have an underlying pathology, therefore the diagnoses of INC has decreased.
After ruling out any aetiology that would require surgery, patients should be directed to medical treatment. Medical treatment should be regulated according to the symptoms and aetiology.
In short, the evaluation and treatment of cholestasis in children, and especially in newborns, is a dynamic process, and the progression of molecular biology and genetic studies will accelerate this.