Abstract
Hepatic encephalopathy (HE) is a reversible syndrome observed in patients with liver disease. The syndrome is characterised by a spectrum of neuropsychiatric abnormalities resulting from the accumulation of neurotoxic substances in the bloodstream and ultimately in the brain. HE is a huge burden to patients, caregivers, and the healthcare system. Common treatments for HE, including rifaximin and lactulose, have been shown to reduce the risk of recurrence, frequency of hospitalisations, hospital costs, and mortality. New research and therapeutics exist, including faecal transplants and small-molecule therapies such as branched-chain amino acids. This review article provides a general overview of the current understanding of HE.
INTRODUCTION
Hepatic encephalopathy (HE) is a complex neurological process that is seen in advanced liver disease. HE causes significant morbidity and mortality and is responsible for considerable burden on patients and their caregivers. HE is treated clinically and can be reversible, especially in the situation of an acute underlying process. This review article will briefly review HE in its entirety: its epidemiology, manifestations, diagnosis, pathophysiology, and treatment as well as future research.
The incidence and prevalence of HE can be difficult to assess because of underlying cause, variable severity of the disease manifestations, and the definition of HE (minimal versus overt).1 Consequently, HE has been reported to occur in a wide range (20–80%) of patients with cirrhosis. The prevalence of overt HE at the time of cirrhosis diagnosis is approximately 10–14%. This rises to 16–21% in patients with decompensated cirrhosis and approximately 10–50% in patients who have undergone transjugular intrahepatic portosystemic shunt (TIPS). Overall, an estimated 30% to 40% of patients with cirrhosis will experience overt HE during their clinical course.1
CLINICAL MANIFESTATIONS
HE can present as a diverse spectrum of neurologic, psychiatric, and musculoskeletal symptoms. In many cases, patients are unaware of the symptoms, but family members or close friends may notice changes and voice their concerns.2 In the early stages, patients may only report subtle symptoms, such as disturbances in their sleep–wake cycles.3 As symptoms progress, patients may develop personality changes such as apathy, irritability, and disinhibition. If left unrecognised, symptoms worsen and patients can present with cognitive impairments such as disorientation, memory impairment, slurred speech, confusion, and eventually coma.4,5 Along with neurologic and psychiatric symptoms, HE can also affect the musculoskeletal system. Patients with minimal HE may show minor issues with coordination, such as changes in their handwriting.6
The most widely recognised symptom of HE is asterixis. However, asterixis is not isolated to HE. It can also be seen in other causes of encephalopathy, such as uraemia.7 Asterixis presents as a flapping tremor that occurs because of negative myoclonus resulting in loss of postural tone.7 It is elicited when patients hyperextend their wrists but can also be observed in the patient’s feet, legs, arms, tongue, and eyelids.6 If it continues to progress, patients can develop hyperreflexia, clonus, and rigidity.6 They can also present with cirrhosis-related Parkinsonism symptoms, with extrapyramidal symptoms including masked facies, rigidity, bradykinesia, slowed speech, and tremors.7,8 These symptoms are seen in approximately 4% of patients, suggesting they are more prevalent than previously thought.8 In patients with TIPS, HE usually becomes clinically apparent 2–3 weeks after TIPS insertion.9
CLASSIFICATION
According to national guidelines, HE is classified using four main axes: the underlying cause, the severity of the disease manifestation, the time course of the disease, and the existence of precipitating factors.6
The first criterion is based on the underlying cause of HE. Type A is HE seen in acute liver failure, Type B in the portal-systemic bypass setting with no intrinsic hepatocellular disease, and Type C is in the setting of cirrhosis with portal hypertension or systemic shunting. The most commonly occurring type is Type C in which patients frequently show signs of liver failure, such as jaundice, ascites, spider telangiectasias, and palmar erythema, among other manifestations.10
The severity of HE is generally classified using the West Haven Criteria (WHC) and the International Society for Hepatic Encephalopathy (ISHEN) criteria (Table 1).6 Alternate grading systems, such as HE Scoring Algorithm (HESA), were developed to provide more reliable grading of HE severity by combining both subjective and objective data.11 Clinical HE Staging Scale (CHESS) is another scoring system that consists of nine clinical items and linearly determines severity using simple terms and questions.12 Various criteria to define the severity of HE are depicted in Table 1.
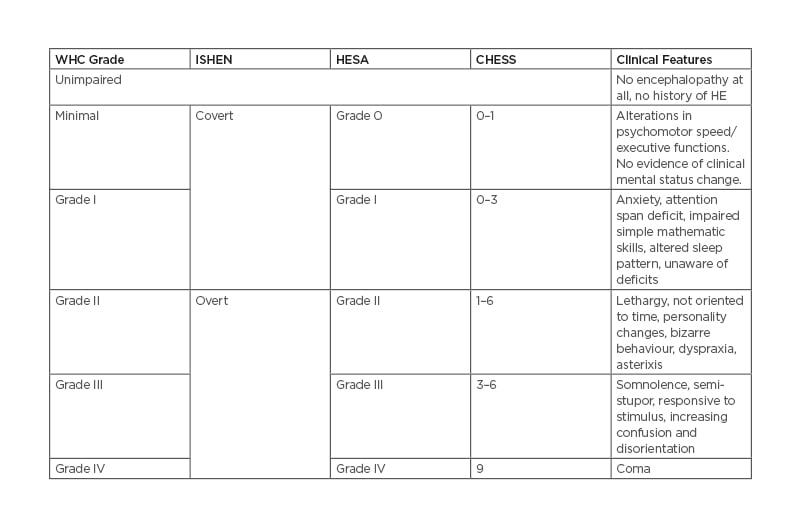
Table 1: Various criteria to define severity of hepatic encephalopathy. The data are reported as mean±SD or median (and interquartile range) as appropriate. HRS: hepatorenal syndrome; MAP: mean arterial pressure: NA, not available.
The third and fourth axes allow patients to be further classified according to the time course and precipitating factors.6 Timing is further sub-classified into episodic, recurrent, or persistent. To classify as recurrent, episodes of HE occur within intervals of less than 6 months, whereas persistent HE involves altered behaviour that is always present in the setting of recurrent HE. Examples of precipitating factors include infections, gastrointestinal bleeding, over-diuresis, electrolyte abnormalities, and constipation. Treating precipitating factors can resolve HE in 90% of all patients.6
DIAGNOSIS
There is no standardised serologic testing or imaging modality to accurately diagnose HE and assess its severity. Ammonia is classically associated with HE; however, increased blood ammonia level alone does not help with the diagnosis or prognosis of HE. Elevation of ammonia in combination with the clinical picture of HE can be more useful, as isolated increased ammonia level can be seen in other medical conditions.13
Current national guidelines suggest the use of an electroencephalogram and complementary neurophysiological tests to diagnose HE in the absence of other neurological process.6 Sensitivity and specificity of an electroencephalogram depend on both modality of data analysis and severity of HE and vary from 57–100% and 41–88%, respectively.14 Neurophysiological tests include psychometric testing of attention as well as working memory and psychomotor speed. Visuospatial ability may also be necessary to identify subtle mental status changes.15,16 Specific examples of these tests and time required to perform them are depicted in Table 2.14,17 While these tests can be beneficial, they are limited because they do not account for results secondary to the patient’s age or baseline education status. They are also limited by time and lack of adequately trained professionals.17
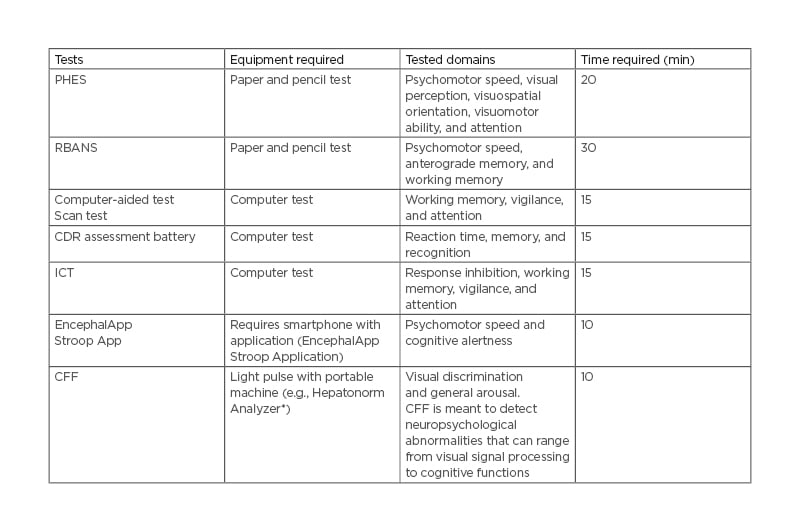
Table 2: Diagnostic and screening methods of hepatic encephalopathy.
* Accelab GmbH, Kusterdingen, Germany. CDR: Cognitive Drug Research; CFF: Critical Flicker Frequency; ICT: Inhibitory Control Test; PHES: Psychometric Hepatic Encephalopathy Score; RBANS: Repeatable Battery for the Assessment of Neuropsychological Status.
Imaging modalities such as CT and MRI scans of the brain are generally non-specific for HE; however, sequential scans may be a useful marker for acute brain volume change.14 PET is not widely used because of cost and limitations of availability; however, PET does provide valuable insight into the pathogenesis of HE in regard to calculating blood flow, glucose metabolism, and ammonia metabolism, which may have prognostic value.14
PATHOPHYSIOLOGY
The pathophysiology of HE is vital in understanding its management. The most well-understood pathophysiological mechanism and correlate of HE is the neurotoxicity of ammonia in the brain, either due to increased production or impaired excretion.18
The two main sites of ammonia production are the small/large intestine (50%) and the kidneys (40%). In the gastrointestinal system, ammonia is produced by degradation of dietary protein to ammonia from urease-producing bacteria as well as breakdown of glutamine by enterocyte glutaminase. Within the kidneys, the proximal tubular cells generate ammonia from glutamine and create bicarbonate as a by-product. The production of ammonia at these sites can be altered through various mechanisms including gastrointestinal bleeding, hypovolaemic states, over-diuresis, hypokalaemia, acidosis, and excessive protein intake.18
The liver is the major site of ammonia catabolism using the urea cycle (Krebs–Henseleit cycle), which converts ammonia into water-soluble urea. The generated urea is subsequently excreted via the intestine and the urine. Due to the reduced capability of the liver to detoxify ammonia, secondary to hepatocellular damage or shunting, ammonia levels increase within the systemic circulation. The kidneys also contribute to decreased ammonia excretion due to acid–base and potassium imbalance, increased protein intake, and dysregulation of glucocorticoid hormones.19 Skeletal muscle also plays a role in ammonia detoxification through glutamine synthase, which converts ammonia to glutamine. Consequently, sarcopenia, a common complication of cirrhosis, can be an adverse factor in HE.20
Dysfunction of the neurons can result from elevations in ammonia in the systemic circulation. Astrocytes, via astrocytic glutamine synthetase, convert ammonia and glutamate into glutamine, which causes increased cerebral volume by osmosis. Consequently, a rapid rise in ammonia can place the patient at risk of brain oedema, as often evidenced on MRI.21 Additionally, glutamine is converted to ammonia in the mitochondria of the astrocytes, which can also lead to direct oxidative damage.22
Another well-described pathophysiological mechanism is alterations in the gut flora that play a vital role in HE. It is theorised that decreased bile acid production allows urease-producing bacteria to proliferate and overtake protective bacteria (i.e., Lachnospiraceae).18,23 Multiple studies have stressed the importance of the microbiota in the development of HE.18,24,25 Several molecules other than ammonia, particularly altered neurotransmitters, may also participate in the development of HE. These can include glutamine, histamine, serotonin, γ-aminobutyric acid, and manganese.18 Further investigation is required to identify additional targets of HE pathophysiology for prompt diagnosis and treatment.
Manifestation of HE is often exacerbated in patients with prior TIPS because of creation of a shunt, allowing portal blood flow to bypass the liver parenchyma. Consequently, ammonia is not broken down or excreted and ultimately crosses the blood–brain barrier, with varying effect on cognitive function.26
TREATMENT
The first-line pharmacologic therapy for HE is laxative. The initial goal of therapy is to prevent ammonia absorption through non-absorbable disaccharides (i.e., lactulose). They are potent laxatives and additionally help alter the intestinal microbiome to non-urease-producing bacteria that reduce ammonia production.27,28 Lactulose also lowers the pH of the colon, causing conversion of ammonia to the ammonium ion, which is not readily absorbable in the colon.29,30 Disaccharide enemas and polyethylene glycol have also been proven to be useful for removing excess ammonia from the colon.29,31 Use of prophylactic therapy to prevent post-TIPS HE was not shown to be beneficial.6,32
Antimicrobials are used to treat HE by targeting culprit gut flora and thereby leading to decreased ammonia production. The most commonly used antimicrobial is rifaximin, a synthetic antibiotic that targets aerobic and anaerobic Gram-negative and Gram-positive bacteria.29,33 Rifaximin has been found to reduce hospital admissions and frequency of recurrent episodes.27,34 In the past, neomycin, vancomycin, and metronidazole have also been used to treat HE but have fallen out of favour.29,35,36
Faecal microbiota transplant (FMT) is a promising treatment for refractory overt HE. Enteric bacteria play an important role in the pathophysiology of HE as described previously. The goal of FMT is to manipulate the gut microbiome in an attempt to reverse intestinal dysbiosis. Two trials exploring the use of FMT in HE found improvement after pre-treatment with antibiotics.29,37,38
Protein restriction is not recommended as normal protein intake levels do not exacerbate or cause HE. Protein restriction may actually be deleterious in patients who are already sarcopenic.27 Other pharmacologic treatments that have efficacy in treating HE are L-ornithine L-aspartate and branched-chain amino acids. L-ornithine L-aspartate works to improve HE by upregulation of two enzymes, carbamoyl phosphate synthetase and glutamine synthetase, which are enzymes for urea and glutamine synthesis; these are impaired in cirrhosis.39 Branched-chain amino acids are theorised to assist with HE through increased detoxification of ammonia in the skeletal muscle. Common treatments for HE are summarised in Table 3.
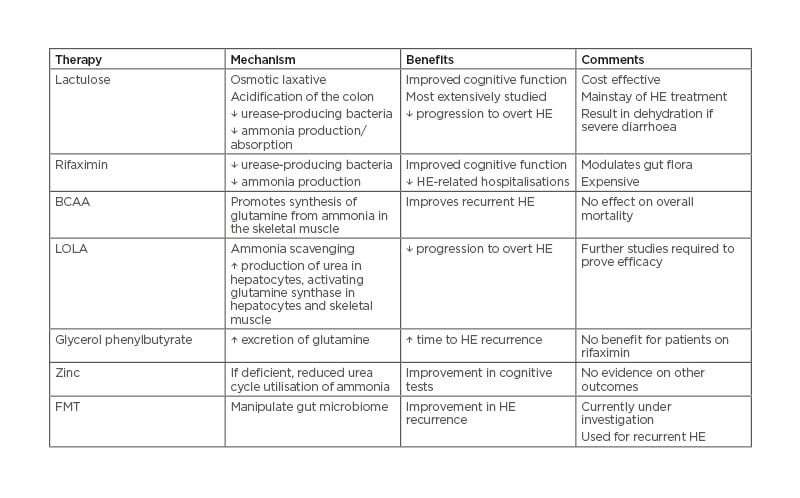
Table 3: Common agents used in the treatment of hepatic encephalopathy. BCAA: branched-chain amino acid; FMT: faecal microbiota transplant; HE: hepatic encephalopathy; LOLA: L-ornithine L-aspartate.
PROGNOSIS
The development of subclinical or overt HE in a patient with liver pathology has been considered one of the adverse markers of decompensation and carries significant morbidity and more. HE was incorporated into early prognostic tools, including the Child–Turcotte–Pugh score, based on clinical experience at the time. The prognosis related to HE varies significantly depending on the decompensation status of the underlying liver cirrhosis.6,40
Subclinical HE has been shown to be present in as many as 80% of patients with cirrhosis in the USA. It may adversely impact activities of daily living, such as driving, which can further affect patients’ socioeconomic status.41
It may adversely impact activities of daily living such as driving; this can further affect patients’ socioeconomic status.42,43 Patients with subclinical HE also have a higher likelihood of developing overt HE within a period of 2 weeks to 2 years after the initial diagnosis.44,45 Further, subclinical HE has been independently associated with increased mortality and the need for liver transplantation, irrespective of the Model for End-Stage Liver Disease (MELD) score.46 Therefore, the diagnosis of subclinical HE is important in prognosticating the development of overt HE as well as overall prognosis.
The development of overt HE is one of the events that defines a decompensated phase of cirrhosis. The development of the first episode of overt HE is independently associated with shorter life expectancies. The cumulative survival of patients who developed overt HE has been found to be <50% in 1 year and <25% at 3 years.47,48 Development of HE has been shown to have a worse 1-year mortality outcome than either variceal bleeding or ascites, which suggests that HE may potentially hold more prognostic value than other events of decompensation.48
The duration of HE, with episodes lasting longer than 48 hours, is important and has been correlated with lower survival rates.49 Higher mortality has also been shown in patients hospitalised with HE and follow-up periods including 28, 90, and 365 days after hospitalisation.50,51 The presence of overt HE is also of prognostic importance in patients with cirrhosis after placement of TIPS. Compared to a single episode of HE, early recurrent overt HE has been found to have three times the increased mortality after adjusting for the MELD score, ascites, albumin, indication for TIPS, and age.52
FUTURE RESEARCH
There are still controversies related to the classification of HE and the role of ammonia measurement in the management of patients with HE.53 Multiple tools are in development to accurately diagnose HE that are not yet validated. The challenge is to keep the clinical diagnostic test simple for the patients while providing high accuracy in diagnosing HE. For example, naming the maximum number of animals displayed in 1 minute has been shown to directly correlate with the grade of encephalopathy.54 A pilot study utilising biomolecular testing has demonstrated the potential use of exhaled volatile organic compounds to detect HE.55 With further improvements in MR and PET technology, research may help personalise prophylactic and therapeutic approaches for patients with HE.53 The classification of HE in the setting of acute-on-chronic liver failure (ACLF) also remains undefined. However, HE in patients with ACLF has distinct clinical features, prognosis, and pathophysiology compared to the conventional forms represented in Types A, B, and C. Further investigations into the classification of HE in patients with ACLF and the role of modulating ammonia in this patient population need to be conducted.50,56
Ultimately, liver transplantation remains the most effective treatment available for cirrhosis and HE; however, it is not always an available option. Unfortunately, permanent cognitive impairment from pre-transplant HE may persist even after this step.
There are a few promising treatments for HE that are still experimental. Probiotics are being evaluated for efficacy in treatment of HE, given the role of microbiota; however, the data that suggests any benefit have been of low quality with no evidence of mortality improvement.29 Minocycline, which reduces microglial cell activation, has been shown to potentially reduce brain oedema and plasma and cerebrospinal fluid ammonia levels.57 Alternative potential treatments include indomethacin, ibuprofen, and phosphodiesterase inhibitors, which restore the function of the glutamate–nitric oxide–cyclic guanine monophosphate pathway in the cerebral cortex in experimental models.29 Benzodiazepine inverse agonists, such as Ro15-4513, have also been shown to be effective in treating HE in animal models of liver failure.58 Flumazenil improved clinical symptoms of HE, albeit temporarily, in a double-blind crossover clinical trial, possibly by decreasing γ-aminobutyric acidergic tone.59 Acetyl-L-carnitine has shown improvement in symptoms of HE as assessed by neuropsychological testing and cognitive abilities, with parallel reductions in serum and brain ammonia levels.60
The use of liver support systems, such as the Molecular Adsorbent Recirculating System (MARS®; Gambro Lundia AB, Lund, Sweden) and Prometheus® device (Fresenius Medical Care, Bad Homburg, Germany), have shown some promise in the acute setting. They function by removing circulating toxins that accumulate in the blood due to liver dysfunction. Both devices have been well tolerated by patients with liver failure and pilot studies suggest improvement in survival rates in some groups of patients with liver failure.61 These devices might have particular utility as bridges to transplantation; however, they might not be appropriate in all patients given the requirement for central venous access and the non-uniform treatment protocols.62
Despite several promising avenues of treatment of HE, studies remain in the early phase and await proof. Future research should understand the impact of comorbidities on outcomes, biomolecular diagnostic strategies, targeting systematic, and neuroinflammation and provide clear endpoints for clinical trials.
CONCLUSION
The clinical presentation of HE may be highly variable and may range from defects in cognition, personality, and intellect, to an altered state of consciousness and impaired neuromuscular function with asterixis and hyperreflexia.
The heterogeneous manifestations of HE vary not only between patients but also longitudinally for an individual patient. By categorising HE’s severity accurately and efficiently, valuable prognostic information and current clinical status can be ascertained.
In conclusion, HE remains one of the most critical prognosticators of worsening cirrhosis, contributing to and correlating with the mortality and morbidity of patients with cirrhosis. Advances in therapeutics are underway to improve clinical outcomes in patients with HE.