
Abstract
Hepatomegaly can be a presenting or associated feature of an inherited lysosomal storage disorder. Thus, a high index of suspicion should be maintained in patients without an aetiologic diagnosis. Suggestive clues can be derived from family history, and/or concomitant manifestations such as anaemia, thrombocytopenia, and splenomegaly. An ophthalmologic examination may disclose relevant findings, such as the presence of a cherry red spot or gaze palsy. Each subtype presents with a broad spectrum of clinical findings, encompassing a wide range of onset from early infancy to adulthood. Diagnostic confirmation necessitates demonstration of decreased enzyme activity and/or mutation in the cognate gene. Diagnosis enables prognostication and consideration of disease-specific therapy, which is currently available for a subset. As inherited traits, diagnosis enables consideration of genetic counselling regarding reproductive risks and implications for relatives. Conditions covered in this review include Gaucher disease, Niemann-Pick diseases, and lysosomal acid lipase deficiency (Wolman disease, cholesterol ester storage disorder).
INTRODUCTION
Hepatomegaly as a clinical problem may represent a manifestation of a storage disorder; that is, a condition resulting from deficiency of an enzyme that would normally metabolise by-products of cellular turnover, and as a consequence tissue deposits build-up in various organs such as the liver, and thus, a characteristic of certain lysosomal storage disorders (LSDs).1 LSDs are relatively rare conditions with broad systemic effects, and associated clinical features help focus evaluation in a patient presenting with hepatomegaly, with or without liver dysfunction (that is, elevated liver transaminases).
Several considerations should raise one’s index of suspicion that a patient’s complaint may be caused by an LSD, while screening for or after preliminary exclusion of more common disorders (e.g., infection, malignancy). LSDs are inherited conditions (mainly autosomal recessive), so obtaining a family history may provide valuable information in cases where there may be more than one similarly affected relative (e.g., sibling). Age of onset, associated clinical manifestations, and a good understanding of clinical disease course also help to direct one’s pursuit of a diagnosis. As diagnostic confirmation requires specialised testing that may not be readily available locally, it may be useful to look at a subset of LSDs, specifically, the sphingolipidoses and related conditions; and look at differentiating one from another with overlapping clinical manifestations, which hopefully leads to a request for the most appropriate investigations.
Herein, the authors describe the hepatic manifestation of three specific LSDs: Gaucher disease (GD), Niemann-Pick diseases (NPD), and lysosomal acid lipase (LAL) deficiency; and their related clinical features, means of diagnostic confirmation, and advances on the therapeutic front.
Of note, the sphingolipidoses (which encompasses Gaucher and NPD A/B) represent a subset of LSDs in which deficiency of a distinct enzyme in the sequential degradation of relevant substrates results in tissue deposits that progressively accumulate in various organs such as the liver and spleen. Except for Fabry disease, which is inherited as an X-linked trait, other conditions within this group are transmitted in an autosomal recessive fashion.2 Incidentally, the classification of LSD was traditionally based on the chemical nature of the substrate that built up, differentiating the sphingolipidoses from another subgroup such as the mucopolysaccharidoses (MPS), characterised in the latter by accumulation in tissues of incompletely metabolised mucopolysaccharides (now referred to as glycosaminoglycans).3 Hepatomegaly can also be found in the MPS, but other features such as the distinctive facial appearance and skeletal changes (i.e., dysostosis multiplex) help to readily distinguish these conditions from the sphingolipidoses and related conditions, the subject of this review (Figure 1).
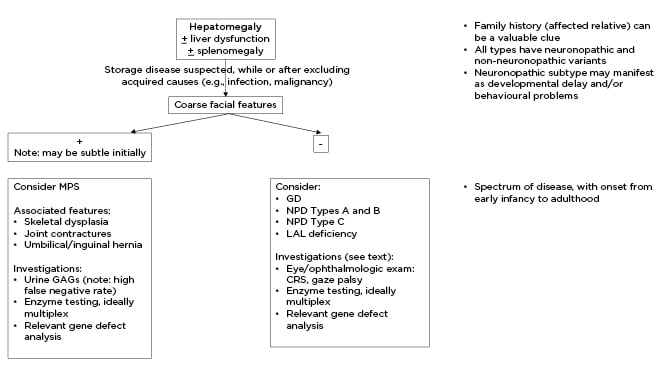
Figure 1: Clinical considerations of lysosomal storage disorders in cases of hepatomegaly, divided into those with and without coarse facial features.
CRS: cherry red spot; GAGs: glycosaminoglycans; GD: Gaucher disease; LAL: lysosomal acid lipase; MPS: mucopolysaccharidosis; NPD: Niemann-Pick disease.
The sphingolipidoses encompass several diseases, besides those reviewed in this paper. The grouping includes Tay-Sachs and Sandhoff disease, GM1 gangliosidoses, Krabbe disease, and metachromatic leukodystrophy. These conditions are dominated by primary neurologic complications in the absence of hepatosplenomegaly. Incidentally, the eponymous designation of several disorders within the group recognises the seminal contribution of the named individual(s), which preceded the identification of the underlying causal defect in all cases. For instance, GD is named after a French physician who described a young female patient who presented with splenomegaly, which on histological examination revealed the characteristic tissue deposits, prior to identification of its biochemical origin.
A related lipidosis, Wolman disease (the severe form of LAL deficiency), is named after Moshe Wolman, an Israeli physician who described the case of three siblings from a consanguineous family with xanthomas in the liver, spleen, adrenals, and other tissues, with death by 3 months of age.4 Subsequently, attenuated cases were described with later onset, lipid deposits in the liver, and hypercholesterolaemia, termed cholesterol ester storage disease.4 With characterisation of the cognate enzyme deficiency, the condition is now preferably called LAL deficiency. As with all the other diseases in this report, it is recognised that there can be a wide age of onset, a broad spectrum of clinical presentation, and variable rate of disease progression. To a certain extent, these observations have been attributed to the presence of residual enzyme activity; thus, a null mutation often leads to an earlier age of onset and more aggressive disease course.
The LSDs associated with hepatomegaly include GD, NPDs, and LAL deficiency. Of these, GD may be the most common, although it is likely that a significant number of patients with NPDs and LAL deficiency are being missed.
Incidentally, the group of diseases referred to as NPD (a designation proposed by Crocker and Farber in 1958) actually encompass different subtypes.5 NPD Types A and B are caused by a deficiency of the lysosomal enzyme acid sphingomyelinase, with Type A representing the severe variant with primary central nervous system (CNS) involvement.5 NPD Types C and D were subsequently shown to be due to defects in two distinct non-catalytic proteins.5 In NPD Type C, 95% of cases result from mutations in a transmembrane protein (NPC1), and the rest arise from mutations in a soluble intra-lysosomal protein (NPC2).6 Historically, NPD Type D referred to affected cases, which are descendants of an Acadian couple who lived in Nova Scotia, Canada, in the early 18th century, a consequence of founder effects (i.e., shared identity by decent).7
NPD Type D has a more homogeneous clinical expression resembling that of less severely affected patients with NPD Type C. Subsequent studies revealed mutations in NPC1 as the cause.
The identification of the molecular basis of several LSDs has prompted the recommendation of an alternative classification. For instance, GD and NPD Types A and B are within a set of designated enzyme deficiency disorders, whilst NPC1 is now grouped with other transmembrane protein deficiency disorders together with defects of sialin (the cause of sialic acid storage disease) and cystinosin (cystinosis).1
CLINICAL PRESENTATIONS
Hepatosplenomegaly is a characteristic feature of GD, although anaemia and thrombocytopenia associated with epistaxis and bruising may be major reasons affected individuals are initially referred for evaluation, in most cases through a haematologist.8 Such may be the case, as liver function is usually preserved in these patients. However, it is recognised that adult patients with GD have an increased risk for gallstones, and it is not unusual that a referral is made first to a gastroenterologist.9 The formation of gallstones has been related to alteration in the composition of bile in the gallbladder, accounting for non-pigment stones (as the anaemia in GD is primarily a consequence of displacement of haematopoietic elements in the bone marrow and sequestration by an enlarged spleen rather than haemolysis). Incidentally, major bleeding does not seem to be a major concern for most patients with GD, as platelet function appears to be largely intact and there is no associated deficiency in clotting factors. However, given the prevalence of GD among individuals of Ashkenazi Jewish ancestry, in whom Factor XI deficiency may also be encountered, bleeding risks may be increased in some cases.10 Regardless, in patients with GD and thrombocytopenia it remains prudent to evaluate patients for bleeding risks prior to any major surgical or dental procedure.
Although liver dysfunction is not a typical feature of GD, cases have been described in patients who have developed cirrhosis; indeed, liver transplantation has been undertaken in a few instances.11 Among patients with GD and parenchymal liver disease, associated complications have included portal hypertension and hepato-pulmonary syndrome.12 Such cases should prompt exclusion of other disease processes, such as infectious or auto-immune hepatitis, and be managed accordingly.
Bone complications, including bone pain and osteonecrosis, are major sources of morbidity in patients with GD; thus, a comprehensive evaluation of disease burden among diagnosed patients must include assessment of bone involvement.13 Suggested bone tests include MRI, bone density, and X-rays of symptomatic sites.
Hepatomegaly is also encountered in patients with NPD. Although in this condition abnormalities in liver function tests (i.e., elevated serum transaminase levels) are often seen as storage material (sphingomyelin), which can be found in hepatic parenchymal cells. This is in contrast to GD, wherein storage (glucosylceramide) is found mainly in Kupffer cells (i.e., cells of monocyte/macrophage lineage).5 There is considerable overlap in clinical features between GD and NPD, except for lung involvement (i.e., interstitial lung disease), which occurs more frequently in the latter.14 Lung involvement (e.g., interstitial lung disease, pulmonary hypertension) can also be encountered in patients with GD, but this problem is seen mainly in splenectomised cases.15
GD and NPD Types A and B are both characterised by a spectrum of features, with primary CNS involvement occurring in a subset. Among patients with GD and CNS involvement are those with an acute (Type 2) and subacute (Type 3) disease course, with death on average before 2 years of age in the former; whilst patients with Type 3 disease live longer (beyond 30 years of age), although often with complications such as seizures.16 Patients with GD Type 3 have a distinctive eye finding attributed to saccadic initiation failure.16 A pertinent negative finding is the absence of a cherry red spot (CRS), characteristic of NPD Type A (also seen in Tay-Sachs disease). The CRS appearance can be accounted for by accumulation of lipids in retinal ganglion cells, with the fovea remaining and appearing unusually red and prominent against the surrounding peri-macular infiltrates. Thus, ophthalmologic investigation can be helpful in the evaluation of patients.17 In NPD Type B, eye findings may be more subtle, described by the presence of a macular ‘halo’.18 In NPD Type C, a characteristic feature is the presence of vertical supranuclear gaze palsy.
LAL deficiency, another LSD characterised by hepatomegaly and liver dysfunction, needs to be considered in patients suspected of having a storage disorder, when the diagnosis of GD or NPD has been excluded. LAL deficiency may manifest as a severe (Wolman disease) or attenuated subtype (cholesterol ester storage disease).4 The Wolman disease variant had been a life-limiting condition, prior to the availability of enzyme therapy. Although there had been reports of patients subjected to haematopoietic stem cell transplantation, it is not certain that significant benefits have been achieved as complications such as sinusoidal obstruction syndrome and progression to cirrhosis have been described in post-transplant cases.20
Non-immune hydrops fetalis (NIHF), a condition characterised by ascites, pleural and/or pericardial effusion, and skin oedema, is a rarely recognised feature of several LSDs, including GD, NPD, and LAL deficiency.21 Other LSDs associated with reported cases of NIHF include sialidosis, galactosialidosis, infantile sialic acid storage disease, MPS Types IV and VII, GM1 gangliosidosis, I-cell disease, and Farber disease. These diagnoses should not be missed when features of NIHF are detected prenatally or at the time of birth, as diagnosis can inform management and also alert couples about recurrence risk with future pregnancies.
DIAGNOSIS
When suspected, the diagnosis of GD, NPD Types A and B, and LAL deficiency can be confirmed by demonstrating deficiency of the cognate enzyme (namely, acid β-glucosidase, acid sphingomyelinase, and LAL, respectively). Increasingly, most laboratories offer multiplex testing, which serves as a quality control measure for samples often shipped to a specialised diagnostic facility.22 Most of these laboratories can now also measure several biomarkers, which may be correlated with disease burden (as discussed below). Note that measurement of enzyme activity does not reliably identify carriers, as values may overlap with results obtained for controls. Relatives at risk may be offered carrier testing by screening for mutations detected in index cases. As NPD Type C is not an enzyme deficiency disorder, diagnosis is confirmed based on characterisation of the causal mutations in either NPC1 (approximately 95% of cases) or NPC2.6 Given the inherited nature of these conditions, affected individuals should be offered genetic counselling regarding reproductive risks and implications for relatives.
A bone marrow biopsy may be performed, given concerns clinical features may suggest a malignancy. In GD, a bone marrow biopsy may reveal the presence of lipid-engorged histocytes with a crumpled silk appearance;23 whilst in NPD, foam cells or sea-blue histiocytes may be observed.24 It should be noted that liquid malignancies associated with high cellular turnover may result in the presence of pseudo-Gaucher cells.25
In jurisdictions where the blood samples may need to be shipped out, waiting for test results may lead to some delay; so, to some extent it may be understandable why a bone marrow biopsy may be done locally to exclude malignancy. However, biochemical and/or molecular testing as noted above is still required for diagnostic confirmation of an LSD, especially prior to introduction of disease-specific therapies (discussed below).
Biomarkers
Several biomarkers, whose concentration or activity can be readily measured in blood, have been identified in patients with a sphinglipidoses and related conditions. However, most of these have been shown to be non-specific and do not necessarily correlate with disease burden (e.g., serum ferritin and angiotensin-converting enzyme in GD). Thus, several investigations have been undertaken to identify biomarkers with greater sensitivity and specificity.
In GD, the current biomarker of choice is lyso-Gb1, with screening studies undertaken in populations at risk to identify affected individuals.26 Chitotriosidase and CCL18/PARC remain popular biomarkers but have less sensitivity and specificity.27 Among affected individuals, studies have shown a correlation with disease burden (e.g., liver and spleen volume, bone involvement).26
The oxysterol species cholestane-3β, 5α, 6β-triol (C-triol) is elevated in patients with NPD Types A, B, and C and LAL deficiency.28 Additionally, lysosphingomyelin and lysosphingomyelin-509 are both elevated in patients with NPD Types A, B, and C, although the magnitude of increase is greater in those with NPD Type C.29
It should be noted that no single biomarker can be fully interpreted in isolation and must be viewed as part of a wider panel of investigations informed by clinical details. Moreover, none of the biomarkers identified to date have full predictive value.
Incidentally, certain results for tests commonly available in most general hospitals may serve as clues to diagnosis. An abnormal lipid profile (elevated low-density lipoprotein cholesterol, low high-density lipoprotein cholesterol) may be found in LAL deficiency, associated with elevated alanine transaminase values.30 A similar profile may be encountered with NPD Types A, B, and C;31 however, lung involvement is not common in patients with LAL deficiency. On the other hand, lung involvement may not be present in the early stages of NPD Types A and B, and thus should not exclude consideration of this diagnosis. The rapid development of neurologic complications in NPD Type A is another important feature associated with the presence of a CRS on eye examination. Neurologic complications are also seen with GD Type 2, which interestingly is not associated with the CRS; as noted, a characteristic finding in Tay-Sachs disease (β-hexosaminidase deficiency), associated with neurologic complications but not with hepatomegaly.32
Of note, epidemiologic investigations have shown individuals with GD have an increased risk for multiple myeloma (MM) and Parkinson’s disease (PD).33,34 With MM, it appears this increased risk may be related to gammopathy as a consequence of chronic B-cell stimulation, wherein the incompletely metabolised lipids act as an antigen and trigger an aberrant immune response.35 In some patients, this drives the clonal proliferation of plasma cells and the development of MM. The basis for the increased risk for PD in GD is under intense scrutiny as it appears the risk is true not only for affected individuals but also for carriers.34 In any case, although the risk for either MM or PD is increased compared to the general population, when looking at a group of patients with GD the proportion of individuals who eventually develop these comorbidities is small. However, at present there is no means for identifying those who go on to develop these problems. Thus, serial monitoring of patients should include screening of serum protein electrophoresis/γ-globulin profile for early detection of MM, and neurologic assessment for features suggestive of PD.
MANAGEMENT
Disease-specific therapies are commercially available for some LSDs and remain investigational for others (Table 1).36
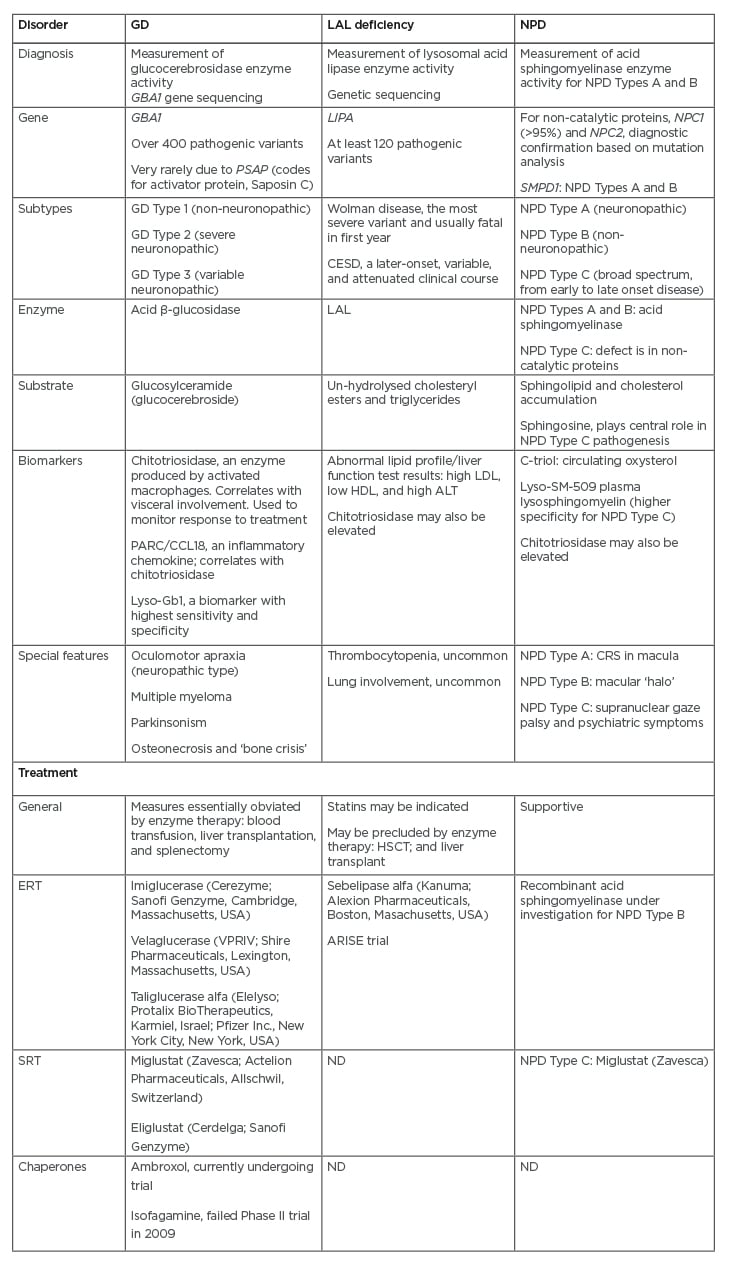
Table 1: Sphingolipidoses associated with hepatomegaly: diagnosis, clinical features, and therapeutic approaches.
ALT: alanine transaminase; CESD: cholesterol ester storage disease; CRS: cherry red spot; ERT: enzyme replacement therapy; GD: Gaucher disease; HDL: high-density lipoproteins; HSCT: haematopoetic stem cell transplant; LAL: lysosomal acid lipase; LDL: low-density lipoproteins; ND: no data; NPD: Niemann-Pick disease SRT: substrate reduction therapy.
Through the years, different approaches have been examined. Those that have achieved regulatory approval include enzyme replacement therapy, the straightforward provision of a recombinant formulation to make up for the deficiency; substrate reduction therapy, the use of a small-molecule agent that inhibits the synthesis of a precursor to limit its build-up in tissues; and pharmacologic chaperone, which is also a small-molecule agent that helps restore the functional conformation of a defective enzyme resulting from misfolding, to spare it from premature degradation and facilitate its delivery to the lysosome.
Enzyme therapy involves the regular intravenous administration of a protein, which has limited access beyond the blood–brain barrier. Antibodies may also form against a recombinant formulation, and when neutralising lead to a loss of efficacy. Small-molecular therapies are administered orally and may be subject to first-pass metabolism; there is also a potential for drug interaction. In the case of pharmacologic chaperones, action may be limited to amenable mutations; that is, gene defects that do not lead to major structural changes (e.g., deletions) that are irreparable.37
Enzyme therapy for GD, which is commercially available, has led to significant improvement in quality of life for treated individuals; however, it does not impact on ultimate neurologic prognosis for those with primary CNS involvement.38
Thus, enzyme therapy is often withheld from those with GD Type 2 but represents the mainstay of therapy for those with Types 1 and 3 disease; in the latter, problems such as seizures are managed symptomatically. As noted, enzyme/protein replacement therapy would not be expected to access the brain, because of the blood–brain barrier; this has prompted investigations of the potential of small-molecule drugs that may either inhibit substrate synthesis or act as a pharmacologic chaperone. Eliglustat (Sanofi Genzyme, Cambridge, Massachusetts, USA), an oral substrate synthesis inhibitor, has been shown to be safe and effective in patients with GD Type 1 naϊve to therapy or switched from a prior regimen of intravenous enzyme therapy.39 Although eliglustat appears to cross the blood–brain barrier, as a P-glycoprotein analogue it is rapidly effluxed; thus, a sufficient drug concentration within the CNS is not achieved. There have been limited investigations involving drugs, such as isofagamine, which act as a pharmacologic chaperone.40 With regards to treatment for CNS involvement, there are ongoing investigations examining the potential of ambroxol and other agents.41
Enzyme therapy is also now available for patients with LAL deficiency, and clinical trials have revealed increased survival in patients with the severe subtypes with salutary changes in lipid profile to a less atherogenic make-up in the attenuated cases, which would be anticipated to reduce long-term complications associated with atherosclerosis (e.g., myocardial infarction, stroke), although this remains to be shown.42 It is likely that early treatment may halt or delay progression to cirrhosis and liver failure in patients with LAL deficiency, but this also needs to be demonstrated. A particular challenge for attenuated subtypes is the variability in disease progression, so long-term follow-up will be required to ascertain the types of patients most likely to benefit and the appropriate time to initiate therapy.
Enzyme therapy for NPD Type B remains investigational, although the outcome of ongoing clinical trials has demonstrated its relative safety and effectiveness in leading to resolution of the systemic manifestations of disease, as observed with enzyme therapy for GD.43
As NPD Type C is not an enzyme deficiency disorder, studies have focused on pharmacologic treatments that may be potentially safe and effective. Miglustat, an orally administered substrate synthesis inhibitor, has been shown to ameliorate some features of disease and improve survival, although responses have not been uniform across patient populations, perhaps because of diagnostic delays prior to treatment initiastion.44 The rationale for the use of miglustat is based on reducing the build-up of substrates, primarily gangliosides. Unlike miglustat, cyclodextrin (2-hydoxypropyl-β-cyclodextrin) does not reduce the rate of substrate synthesis but it has been found to mobilise cholesterol by mechanisms not fully understood. When administered to the mouse and cat models of NPD Type C1, cyclodextrin was shown to extend life, reduce cerebellar pathology, and delay the onset of ataxia.45 Clinical trials with cyclodextrin, administered either intravenously or intraventricularly, are underway.46 Another drug under consideration is arimoclomol, a small-molecule co-inducer of heat shock proteins that is currently in clinical trials for NPD Type C; in vitro studies involving fibroblasts obtained from affected individuals have shown recombinant human heat shock protein 70 improved the binding of several sphingolipid-degrading enzymes to their essential co-factor bis(monoacyl)glycerophosphate, and the potential for reducing tissue substrate storage.47
In patients with LAL deficiency, the use of lipid-lowering drugs has been considered as a means of reducing cardiovascular comorbidity.48 However, its use may not obviate the need for enzyme therapy as such an approach would not lead to eliminating damage to the liver, which can develop in affected individuals. Regardless, it raises the prospect of combination therapy to promote an optimal outcome, but this requires closer examination.
CONCLUSION
In patients, whether children or adults, presenting with hepatomegaly as a finding or chief complaint, a high index of suspicion should be maintained that the clinical problem may be caused by a lysosomal storage disease. A positive family history for similar manifestations may be a valuable clue, given the conditions described (namely, GD, NPDs, and LAL deficiency) are all inherited autosomal recessive traits. However, the absence of a family history should not exclude diagnostic consideration and investigations should continue when other clinical manifestations are suggestive (e.g., presence of anaemia, thrombocytopenia, splenomegaly, lung involvement, and/or bone disease). Neurologic involvement may also be encountered in a subset of these conditions.
Diagnostic confirmation in these instances is established based on demonstration of the underlying enzyme deficiency, complemented by molecular genetic testing. An exception is with NPD Type C, as the underlying cause lies with either a gene defect encoding a transmembrane or soluble non-catalytic protein; thus, molecular genetic testing is required. Changes in concentrations of several biomarkers may provide additional clues, but as with diagnostic confirmation the availability of such testing may be limited to specialised laboratories. For these sphingolipidoses and related conditions, carrier screening can be performed reliably by genetic testing, when the causal mutation segregating in the family is identified.
Enzyme therapy is available for GD and LAL deficiency and under investigation for NPD Type B, whilst therapeutic trials for NPD Type C explore different pharmacologic options. Early diagnosis is key to achieving a favourable clinical outcome. Of recent interest are comorbidities that have become a focus of research, as they have been recognised as part of perhaps an expanded spectrum of disease, now that the cardinal features have been effectively managed by therapeutic options (e.g., MM and PD in GD). Despite their rarity, valuable lessons have been learned as we await the promise of gene therapy. A principal unmet need is treatment for the CNS manifestations.





