Abstract
Hepatopulmonary syndrome (HPS) may be defined by hepatic disease, gas exchange abnormalities that may lead to hypoxaemia, and the presence of pulmonary vascular dilations. The balance between the many substances involved in vasodilation and vasoconstriction is regulated by the liver; thus, liver damage may generate systemic changes throughout the body. The pulmonary tissue may be damaged by reactive oxygen species or nitric oxide. Dyspnoea is the most frequent pulmonary symptom, caused by tissue damage, and may become worse when an individual exercises. In experimental research the surgical model of bile duct ligation is the optimal model to simulate the typical lung alterations present in HPS, which results in an increase in oxidative stress in hepatic and pulmonary tissues. In liver injury, the muscular system may also be damaged, for example sarcopenia may seriously aggravate cirrhosis and is associated with cirrhotic patient mortality. Muscular changes can be explained by the actions of myostatin and insulin-like growth factor and the increase in body levels of ammonia. As a result of impaired cardiopulmonary and muscular conditions, HPS patients may exhibit a low exercise tolerance, low muscle strength, and low functionality. Liver disease can contribute to HPS oxidative stress and is one of the main factors responsible for the reduction of gas exchange. Physical exercise can be performed as a way of modifying this pathophysiological state. Studies that have investigated physical exercise as a therapy for cirrhosis suggest that this approach may be beneficial for cirrhotic patients, primarily with regard to muscular and cardiorespiratory injuries.
INTRODUCTION
Hepatopulmonary syndrome (HPS) may be defined by hepatic disease, gas exchange abnormalities that may lead to hypoxaemia, and the presence of pulmonary vascular dilations.1 The physiopathology of the hypoxaemia in HPS is multifactorial and involves intrapulmonary shunting, a low ventilation-perfusion ratio, low diffusion, and an unbalanced alveolar-arterial oxygen gradient (AaDO2). The prevalence of HPS in cirrhotic patients is approximately 10–20% and contributes to an increased morbidity and mortality in these individuals.1,2
Hypoxaemic events are common in patients with HPS, and these alterations may be caused by intrapulmonary vascular dilations associated with hepatic disease. Lung capillary dilation is a typical vascular alteration of HPS; it may be localised and is less frequently present in the pleural and arteriovenous lung communications.3 The hypoxaemia in these situations may be explained by the unbalance of the ventilation-perfusion ratio, the presence of intrapulmonary shunts, and the decrease in the diffusion caused by intrapulmonary vasodilation.4,5
Vascular disorganisation results in the appearance of new alveolar units. These units have preserved ventilation; however, the presence of an arteriovenous shunt and intrapulmonary vasodilation compromises the maintenance of proper levels of arterial oxygen. The presence of biomolecular mediators produced by the liver, such as nitric oxide (NO), which is a powerful biological vasodilator derived from the endothelium, may be the main factor causing pulmonary vasodilation. Decreased oxygen flow to the blood capillaries does not occur because of the thickening of the alveoli-capillary membrane, it occurs because of poor vascular dilation, which is associated with increased cardiac output, a common alteration of cirrhotic patients with hyperdynamic circulation.6-8 Liver disease can contribute to HPS, where oxidative stress is one of the main factors responsible for the reduction of gas exchange, and physical exercise can be performed as a way of modifying this pathophysiological state.
DIAGNOSIS, CLASSIFICATION, AND SYMPTOMS
The main features present in HPS patients are dyspnoea and platypnoea, (with an increase in dyspnoea when changing from lying to standing), which may be associated with fatigue, as well as the clinical findings of peripheral cyanosis and digital clubbing. Dyspnoea is considered the more frequent pulmonary symptom and may worsen when the patient exercises.6 The diagnosis is made when the AaDO2 is >20 mmHg or when there is hypoxaemia, which is defined by a partial pressure of oxygen (PaO2) <70 mmHg. Intravascular lung dilations may be present, which facilitates confirmation of the diagnosis made by an echocardiogram.8
According to Krowka and Cortese,9 HPS may be classified into two subtypes based on the PaO2 and angiograph findings. HPS type I is defined by the presence of precapillary diffuse dilations that respond well to oxygen therapy. HPS Type II is characterised by small vasodilatation and arteriovenous malformation. In contrast, Rodríguez-Roisin et al.1 suggest a staging classification of HPS into four groups based on the gas exchange abnormalities, which are considered mild when the PaO2 is >80 mmHg, moderate when the PaO2 is 60–80 mmHg, severe when the PaO2 is 50–60 mmHg, and very severe when it is <50 mmHg.
HEPATIC DISEASE AND OXIDATIVE STRESS
The liver is critical in regulating the balance between vasoconstrictor and vasodilator substances, and hepatic tissue injury can impair the regulatory functions that can lead to many abnormalities. Liver damage makes it more difficult for the organ to filter blood from the portal vein, which leads to the appearance of portosystemic shunts and a decrease in the hepatic phagocytic capacity. As a result, the lung filtrates systemic blood to compensate for the decrease in hepatic phagocytosis,10 and the increase in the lung phagocytic activity results in macrophage accumulation in the pulmonary endothelium and increases cytokine and NO levels in the extracellular environment. NO acts as a molecular signal that mediates the vasodilation of pulmonary capillaries when it is produced in low concentrations by vascular endothelial cells. NO has a toxic oxidant action when it is produced in high concentrations by lung macrophages. Phagocytes also produce the superoxide anion radical, which oxides the plasmatic membrane and produces a substantial amount of reactive oxygen species (ROS).11
ROS and reactive nitrogen species are products of physiological cellular metabolism and have both noxious and beneficial effects on the organism. Many physiological functions are controlled by these molecules, including vascular tonus regulation, defence against invading micro-organisms and harmful cells, and monitoring of oxygen pressure in ventilation control and molecular levels.12 An unbalance in this system can lead to tissue damage and can result in chronic-degenerative diseases.13
Tieppo et al.14 assessed the lipoperoxidation of pulmonary tissue following the experimental model of common bile duct ligation (BDL); using the techniques of thiobarbituric acid reactive substances and chemiluminescence, the authors identified a significant increase in the lipid peroxidation, which may be explained by the action of phagocytic cells when fighting against the process of bacterial translocation. The migration of macrophages to the lungs leads to the production of the superoxide anion radical and increases the antioxidant enzyme activity. Tieppo et al.14 identified a significant increase in the enzyme superoxide dismutase in the pulmonary tissue of the cirrhotic group, which suggests increased enzyme activity as a result of the aggression induced by ROS. The researchers concluded that the experimental model of common BDL simulates the main symptoms of HPS, which suggests it is a good model to investigate oxidative stress, hepatic changes, and the respiratory system.
HEPATOPULMONARY SYNDROME AND EXPERIMENTAL MODELS
The experimental models more commonly used to investigate the abnormalities caused by HPS are the BDL and partial portal vein ligation in rats. In both models, NO develops an important rule regarding the pathogenesis of the experimental HPS, which causes vascular dilation and leads to hypoxaemia, similar to the findings of cirrhotic patients.
Experimental studies that used the BDL surgical model have indicated an increase in oxidative stress of hepatic and pulmonary tissues, as well as other lung alterations typical of HPS.14 Long-term bile duct obstruction causes secondary biliary cirrhosis, which leads to acute inflammatory reactions, causing systemic changes.15 Chang and Ohara16 demonstrated that animals that had undergone a BDL surgery presented progressive worsening of the gas exchange, which is similar to the changes that occur in humans with HPS. The researchers determined that the cirrhotic animals had an AaDO2 >100 mmHg, associated with moderate hypoxaemia (values <85 mmHg). These findings may be linked with intrapulmonary vasodilation caused by high levels of endothelial NO synthase present in pulmonary tissues.17
Tieppo et al.14 identified a decrease in the PaO2 and peripheral oxygen saturation in haemoglobin, and a significant increase in the partial pressure of carbon dioxide in the arterial blood in cirrhotic animals compared with the control group. The same researchers also identified a significant difference in the AaDO2 between the BDL and sham groups, which is consistent with the findings of Luo et al.18 that showed animals developed progressive hyperventilation and hypoxaemia in association with the increase in the AaDO2.
Vercelino et al.19 suggested that the experimental model of BDL best simulates the alterations present in HPS. The researchers investigated the gas exchange abnormalities of different experimental models of hepatic cirrhosis (Table 1), and the BDL model exhibited more changes in the gas exchange values. An increase in partial pressure of carbon dioxide in the arterial blood was identified in the BDL group compared with the control group, as well as alterations in the PaO2, arterial oxygen saturation, and AaDO2. In addition to the gas exchange abnormalities, the BDL group also exhibited a larger intrapulmonary vasodilation evidenced by histological analysis. Therefore, BDL is a method proven to be efficient at simulating vascular alterations and gas exchange abnormalities identified in HPS.
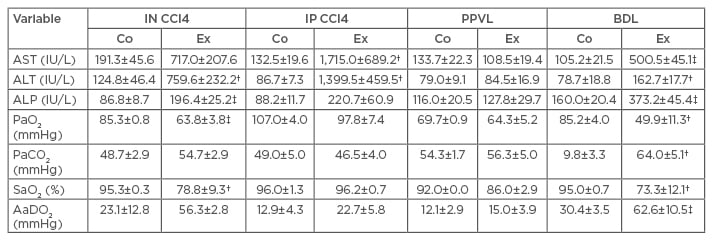
Table 1: Serum enzyme levels, blood gases, and alveolar-arterial oxygen gradient in control and experimental groups of the four experimental models of cirrhosis.*
*The results expressed as the means ± standard errors of the means; †p<0.05 versus the corresponding control group; ‡p<0.001 versus the corresponding control group.
AaDO2: alveolar-arterial oxygen gradient; ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; BDL: bile duct ligation; Co: control; Ex: experimental; IN CCl4: inhaled carbon tetrachloride; IP CCl4: intraperitoneal carbon tetrachloride; PaO2: arterial oxygen tension; PaCO2: arterial carbon dioxide tension; PPVL: partial portal vein ligation; SaO2: arterial oxygen saturation.
Berthelot et al.20 identified an increase in the diameter of the lung arterioles of animals that had undergone BDL surgery, in which the ratio between the pulmonary weight and body weight increased in the cirrhotic group compared with the control group. These alterations are likely caused by pulmonary shunts and intrapulmonary vascular dilations, as a result of intrapulmonary capillary congestion. The intrapulmonary vasodilation in these cases may be associated with bacterial translocation, which occurs as a result of the absence or decrease of bile in the small intestine. This bile deficit decreases the emulsifier and antiendotoxic effect of the bile salts, which leads to an increase in the endotoxin levels in the large intestine, which are carried by the portal circulation to the pulmonary system.21
MUSCULOSKELETAL ALTERATIONS IN HEPATOPULMONARY SYNDROME
Striated muscle is a complex tissue that is versatile, heterogeneous, and composed of specialised multinucleate muscle fibres. These cells have a myonuclei placed in the periphery of the fibre immediately below the plasmatic membrane. Contractile units, also referred to as sarcomeres, are morphologically repeated and are comprised of muscle fibres. Each sarcomere is composed of many proteins, including the contractile proteins of myosin, a thick filament formed by the polymerisation of 200–300 molecules of Class II myosin, and actin, a thin filament associated with the regulator proteins of troponin and tropomyosin.1
Similar to other chronic diseases, the muscular system is one of the most damaged in hepatic injuries and determines the patient’s limitations regarding physical exercise. Striated muscle makes up 45% of the body weight, and is critical in the preservation of bioenergetics, homeostasis, once it is the main place of processing, and storage of energy, as well as being the final destination of the primary support systems involved in exercising, such as the cardiovascular and respiratory systems.1 Sarcopenia, or muscle loss, is the most common muscle abnormality of liver diseases and only a limited number of studies have investigated the mechanisms involved in this process.22 In cirrhosis, sarcopenia may aggravate the clinical condition of patients because it decreases functionality, contributes to a worse disease prognosis, and, as an independent factor, increases the mortality of the cirrhotic population.23 Malnutrition in cirrhotic patients is also associated with a worse disease prognosis; when combined with low food intake and high metabolic activity, malnutrition may lead to cachexia, damaging the muscular system.24
Sarcopenia is the result of a reduction in protein synthesis and an increase in muscular proteolysis, as well as the proliferation and uncontrolled differentiation of satellite cells of the muscular system.25 Muscular tissue alterations depend on molecular factors, such as myostatin and insulin-like growth factor 1, which are the main regulators of the muscle skeletal growth process.26 Myostatin and insulin-like growth factor 1 also participate in protein synthesis regulation and an increase in proteolysis, with high levels of myostatin identified in cirrhotic patients with end-stage liver disease.26,27
Many other molecular mediators also contribute to sarcopenia in cirrhotic patients, with an excess of serum ammonia levels one of the main factors involved in this process.28 Once the liver is the main organ responsible for controlling ammonia levels in the body, the presence of hepatic dysfunction results in increased serum ammonia levels.28 Hyperammonaemia results in a process referred to as nitration, which damages protein cell functioning when the serum ammonia levels are increased.28 In an attempt to degrade the damaged proteins of the skeletal muscle, a process of cellular autophagy may be initiated; however, the autophagy process may not work properly in specific diseases28 and is associated with tissue damage in hepatic cirrhosis.
In contrast to sarcopenia, cachexia is defined as a concomitant loss of muscle and fat tissues.22 Cachexia has also been associated with malnutrition, which is intensified by the high resting energy demand and is not supplied because of an inadequate absorption of nutrients in cirrhotic patients.24 Furthermore, the physiological state of hypermetabolism in association with bacterial translocation episodes activates the production of proinflammatory cytokines in cirrhotic patients, leading to protein catabolism and decreases nutrient absorption.24
Figure 1, modified from Dasarathy22 and Córdoba and Mínguez,29 shows the increase in the serum ammonia levels as a result of hepatocellular damage. Ammonia may damage astrocytes when it crosses the blood–brain barrier, thereby damaging mitochondria and the transport and metabolism of glutamate and glutamine. The presence of ammonia in the systemic circulation may also damage the muscular tissue, thereby interfering in protein synthesis and causing sarcopenia. Physical exercise and the use of myostatin antagonists appear to be potential therapeutic approaches to revert sarcopenia, as well as leucine supplementation, once there is a low level of available amino acids in the body of patients with hepatic dysfunction.
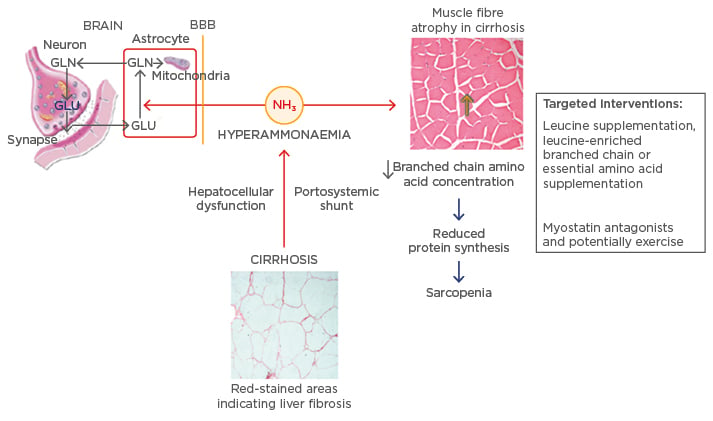
Figure 1: Alterations caused by ammonia in cirrhosis and its targeted interventions.
BBB: blood brain barrier; GLU: glutamate; GLN: glutamine.
Adapted from Dasarathy22 and Córdoba and Mínguez.29
PHYSICAL EXERCISE AND LIVER DISEASE
Gas exchange abnormalities are associated with cirrhosis and are evident in HPS patients, which, thus, influence their exercise tolerance. HPS patients exhibit a low maximal oxygen consumption (VO2max), worse results in the 6 minute walk test, and lower respiratory muscular strength compared with cirrhotic patients who do not have an HPS diagnosis.30 Furthermore, cirrhotic patients who have an increased AaDO2 have earlier ventilatory thresholds and worse gas exchange during maximal efforts.31
The muscular and respiratory weakness of cirrhotic patients negatively affects their exercise tolerance and functionality. Andersen et al.32 stated that patients with alcoholic cirrhosis have decreased lower and upper limb strength compared with healthy subjects. Tarter et al.33 have also demonstrated that cirrhotic patients have lower strength capacities of the lower and upper limbs when both concentric and eccentric contractions were assessed. Moreover, the decrease in peripheral muscle strength may affect the respiratory muscle strength. Kaltsakas et al.34 and Abdel-bary et al.35 identified a negative correlation between the respiratory muscle strength and dyspnoea in cirrhotic patients. Galant et al.36 identified a correlation between the VO2max and respiratory strength and a decrease in the quality of life, which suggests an interaction between physiological factors and decreased functionality in cirrhotic patients.
Different studies that investigated physical exercise as a therapeutic approach for cirrhosis suggest that these patients may benefit from exercise.37-39 Child–Pugh Class A and B patients underwent an 8-week aerobic exercise programme on a cycle ergometer, which indicated an increase in the quadriceps mass via ultrasound analysis and VO2max improvement.37 Another study, which included cirrhotic patients from a waiting list for liver transplantation, demonstrated improvements in VO2max, knee extensor maximal strength, and 6-minute walk distance following a 12-week programme of aerobic and strength exercises.38 Furthermore, cirrhotic patients with a model for end-stage liver disease <25 improved their 6-minute walk distance, step test values, and muscle mass following a 12-week moderate exercise programme on a treadmill and bicycle, which was associated with leucine supplementation.39
CONCLUSION
Physiological alterations present in HPS patients are similar to the alterations present in experimental models of BDL. The clinical features of intrapulmonary vasodilation and gas exchange abnormalities are better understood when investigated in combination with the antioxidant system, inflammatory processes associated with bacterial translocation, and the presence of portosystemic and intrapulmonary shunts. HPS, and other diseases that affect the hepatic system, may contribute to the development of sarcopenia and cachexia because of hyperammonaemia, malnutrition, hypermetabolism, and proteolysis. Physical exercise is a potential therapeutic approach to revert existing impairments in the musculoskeletal and pulmonary systems and is an option for the treatment of HPS patients and other individuals with hepatic injury.