Abstract
Nonalcoholic steatohepatitis (NASH) is a subtype of nonalcoholic fatty liver disease that is characterised by steatosis, chronic inflammation, and hepatocellular injury with or without fibrosis. The role and activation of macrophages in the pathogenesis of NASH is complex and is being studied for possible therapeutic options to help the millions of people diagnosed with the disease. The purpose of this review is to discuss the pathogenesis of NASH through the activation and role of Kupffer cells and other macrophages in causing inflammation and progression of NASH. Furthermore, this review aims to outline some of the current therapeutic options targeting the pathogenesis of NASH.
INTRODUCTION
Nonalcoholic steatohepatitis (NASH), a subtype of nonalcoholic fatty liver disease (NAFLD), is one of the most prevalent ongoing liver diseases seen across the world. NAFLD is histologically divided into two types: nonalcoholic fatty liver and NASH. There is an estimated 20–40% prevalence of NAFLD worldwide and approximately 10–20% of those affected progress to the subtype NASH.1 NASH is histologically characterised by the accumulation of dense lipid deposits in hepatocytes, causing inflammation and hepatic cell injury. This injury leads to hepatic fibrosis, the chief cause of hepatic and extrahepatic complications.
The progression to NASH from its less severe form of NAFLD can be predicted by the amount of inflammation present in hepatic tissue.2 Severe inflammation can contribute to the progression of other liver diseases, such as cirrhosis, fibrosis, and hepatocellular carcinoma.
The cells’ first line of defence against hepatic cell injury is the activation of macrophages. The increased activation of inflammatory macrophages produces inflammatory cytokines, which determine the progress of NASH. In this review, we aim to understand the role of the resident and infiltrating macrophages present in NASH. Additionally, we intend to summarise potential mechanisms and future therapeutic options that aim to reduce the burden of macrophages in NASH.
KUPFFER CELLS
Abundancy
The liver has an abundance of macrophages that spur the development of NASH by means of extensive inflammatory pathways resulting from activated macrophages. It is estimated that for every 100 hepatocytes, there are an additional 20–40 macrophages supplementing the hepatocytes.3 The majority of macrophages present in liver tissue are the self-renewing, resident phagocytic Kupffer cells (KC). These are split into M1 and M2 subsets that, in a healthy liver, balance each other’s functions.
Topology
KC reside in liver sinusoids, the portal tract, and hepatic lymph nodes at the crossroads of capillary-level confluence of the portal vein and hepatic artery tributaries. At that junction they are in an environment that contains various inflammatory agents as a result of hepatic circulation. KC distribution within the liver acinus is correlated with the acinar concentration gradient of immune reactive substrates and other regulatory factors.
Functionality
The main function of KC is to detect and destroy pathogens, cell debris, and bacterial-derived products in the hepatic circulation by phagocytosis, preventing the general circulation of such pathogens. In healthy livers, KC fulfill the dual function of clearing these pathogens while keeping a low and balanced level of inflammation. KC use microbe-associated molecular pathways to bind microbes or microbe ligands. KC function via pattern recognition receptors (PRR) that can be divided into two classes: toll-like receptors (TLR) and NOD-like receptors (NLR).4 These receptors detect danger signals, including pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP), which leads to the activation of inflammatory pathways. KC are then responsible for clearing these microbes via phagocytosis to prevent them from penetrating general circulation.5 Additionally, KC may be activated by metabolically driven activated signals.6
KC clear microbes whilst keeping the hepatic area at an optimally controlled level of inflammation. This protects the rest of the body from an excessive immune response. KC produce and secrete anti-inflammatory signals to respond to lipotoxicity, including interleukin (IL)-10 in response to lipopolysaccharide (LPS).7 In this manner, a balanced response is produced by KC. Additionally, KC participate in immunosuppression by expressing high amounts of T cell suppression molecules and low levels of costimulatory molecules.8
In conclusion, KC maintain homeostasis and govern inflammation in the liver microenvironment.
Subtypes
There are two main subsets of inflammatory macrophages that are separated based upon their terminal differentiation stage: the proinflammatory M1 and immunoregulatory M2 macrophages. These macrophages perform multiple functions, such as cytokine and chemokine secretion, leukocyte adhesion, phagocytosis, and cellular crosstalk. Later studies9,10 have shown that the M2 type expands to include many other macrophages with vast differences in their biochemistry and physiology, leading to a classification system based on a full spectrum. M2 macrophages have been subdivided into M2a, M2b, and M2c, each with different regulators, marker proteins, and special functional activity.9 M1 macrophages are characterised by expression of high levels of proinflammatory cytokines, reactive intermediates, and promotion of a T helper cell (Th)1 response. M2 macrophages exhibit phagocytic activity, tissue remodelling, and tumour progression. A more recent study10 has shown that the balance of M1 and M2 macrophages regulates inflammation in the liver and is the underlying factor in NASH when the levels of each macrophage subtype are unbalanced (Figure 1).
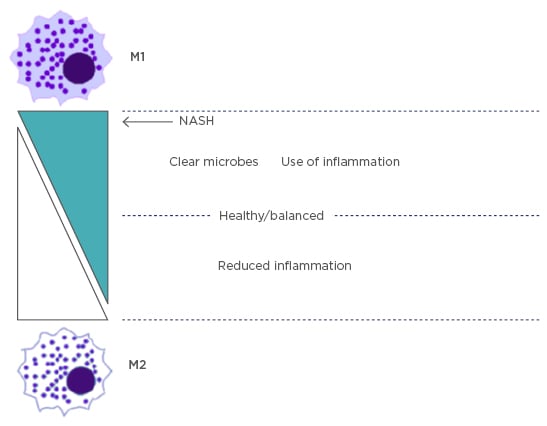
Figure 1: The balance of M1 and M2 macrophage subtypes.
Kupffer cells are in balance between M1 and M2 subtypes to control inflammation. In NASH, the key marker is an imbalance of M1 causing excess inflammation.
NASH: nonalcoholic steatohepatitis.
Activation of M2 Type
M2 macrophages are primarily responsible for wound healing and exhibit anti-inflammatory properties. M2 macrophages are induced and activated by IL-4, IL-10, IL-13, IL-33, tumour growth factor (TGF)-α, TGF-β, peroxisome proliferator-activated receptors (PPAR)-γ, and possibly PPAR-δ.11-13
IL-4 has been shown to promote the expansion of M2 macrophages by initiating Th2 differentiation and downregulating production of proinflammatory chemotactic factors.14,15 IL-4 is also an inducer of endogenous PPAR-γ ligands.13 IL-10 downregulates Th1 cytokine expression and suppresses antigen presentation. IL-13 induces secretion of TGF-β and allows for alternative macrophage activation, allergic inflammation, and immunoglobulin (Ig)E secretion.
PPAR-γ primarily controls the expression of gene networks involved in adipogenesis, lipid metabolism, inflammation, and the maintenance of metabolic homeostasis. Activation of PPAR-γ inhibits inflammatory gene expression by preventing the inflammatory signal-specific removal of the corepressor complex.16 In addition, studies have shown that PPAR-δ regulates an anti-inflammatory switch that proceeds through a ligand activation and genetic receptor depletion.17 Although PPAR-δ has been shown to promote mouse M2 macrophages, it is still not completely known whether PPAR-δ signals and functions in activation of human M2 macrophages.18 Studies on human M2 macrophages were performed on atherosclerotic lesions and may give different results in NASH-derived hepatic cells.
M2 macrophages produce anti-inflammatory and profibrotic cytokines. In response to IL-4 and IL-13, M2 macrophages promote Th2 responses.9 Additionally, M2 macrophages express high levels of arginase, which promotes anti-inflammatory responses and increases mannose receptor expression (Figure 2).19
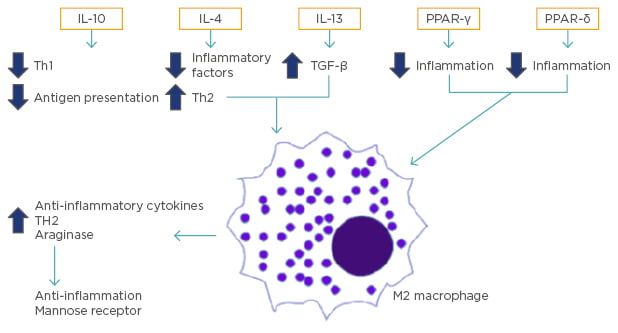
Figure 2: Activation and promotion of M2 type macrophages.
The activation of M2 macrophages is caused by cell signals, including IL-10, IL-4, IL-13, PPAR-γ, and perhaps PPAR-δ. These lead to the reduction of inflammatory factors, downregulation of Th1 cytokine expression, suppression of antigen presentation, and alternative macrophage activation, which ultimately leads to the production of antiinflammatory cytokines and profibrotic cytokines.
IL: interleukin; PPAR: peroxisome proliferator-activated receptors; TGF: tumour growth factor; Th: T helper cell.
Activation of M1 Type
In NASH, KC are the first macrophages to be activated and thus are of critical importance in the progression of NAFLD.19 The classically activated macrophages, the M1 type, promote inflammation. They are induced by proinflammatory signals such as interferon (IFN)-γ, tumour necrosis factor (TNF), and LPS that are present in high fat diets.18 In NAFLD, LPS levels are raised in portal circulation due to dietary factors. High fructose diets regress NAFLD due to the increase of bacterial levels and intestinal permeability. Disruption of the liver mucosal barrier will allow PAMP to bind to PRR and activate immune cells. Depletion of KC have been shown to protect against the development of steatosis.20
Microvesicles released by fat-laden cells undergoing lipotoxicity contribute to the activation of M1 macrophages. It has been suggested that these microvesicles can activate the NLRP3 inflammasome following internalisation by macrophages.21 In recognising inflammatory substances, KC utilise PRR, including TLR. The various TLR allow for recognition of different microbial products. TLR2 recognises peptidoglycan and results in the release of proinflammatory cytokines,22 TLR4 recognises LPS, and TLR9 recognises foreign nucleic acids.4,23 Additionally, toxic lipids stimulate TLR to respond to LPS.20
LPS has been shown to activate KC by binding to TLR4 and its associated protein, CD14, as well as myeloid differentiation-2 molecule, thereby activating a cascade of inflammatory signalling pathways.24 Free fatty acids have also been shown to act on TLR4 via a supplemental ligand.25 TLR4 deprived mice exhibited less severe hepatic injury and less hepatic lipid accumulation, thus placing TLR4 as an essential mediator in inflammation processes and NASH.26 Studies in which TLR9-deficient mice were fed a choline-deficient amino acid-defined diet showed less severe hepatic injury and those studies have linked TLR9 signalling to inflammasome activation.27
PROGRESSION OF NONALCOHOLIC STEATOHEPATITIS
The hallmark of NAFLD is an excess of fatty acids and lipids in the liver that results in lipotoxicity and hepatocyte injury that initiates inflammation.28 The immune system attempts to recover from this inflammation through the release of cytokines from KC; thus, unintentionally furthering inflammation by activating other pathways, leading to steatosis and NASH. Recently, it has been demonstrated that a true lipid signature of NASH exists and is seen spreading into the hepatic parenchyma of selectively accumulated fatty acids.29 This is caused by a change in the metabolic pathway involved in the synthesis of long-chain fatty acids and very long-chain fatty acids.29
Previously, a ‘two-hit model’ was proposed as the pathogenesis mechanism of NASH. The metabolic syndrome involving triglyceride accumulation is the first hit, and the second hit is defined as the progression to liver inflammation, oxidation, and progression to steatohepatitis via KC.30 More recently, studies have shown that inflammatory mediators, based on the activation of KC and the release of cytokines, play a central role in the cascade of inflammation and liver injury, and that the inflammation may precede the development of steatosis.31 The amount of data implicating gut microbiota and genetic factors has led to the development of the ‘multiple parallel hits model’ that accounts for the observed cases of NASH, even in lean subjects.32
In addition, hepatocyte cell death is a key process in the pathogenesis of NASH.28 Death receptors have been shown to mediate inflammatory signalling.32 Cells undergoing necrosis and apoptosis release DAMP, which further induces inflammation by activating inflammasomes such as NLR proteins (NLRP). The DAMP associated with M1 macrophages are high motility group box 1, heat shock proteins, breakdown products of extracellular matrix, and nonprotein substrates.33 The activation of NLRP can cause the assembly of the inflammasome, which contains caspase-1, causing further inflammation and cell death by cleaving prointerleukins into their interleukin form.34 These inflammasomes are important in the progression to NASH because this cycle can lead to a full inflammatory response, which can result in fibrosis and cirrhosis.
Apoptosis is upregulated in hepatic cells as a result of harmful diets. Hepatic saturated fatty acids found in adipose tissue can be released from lipid droplets via macrolipophagy. In high areas of saturated fatty acids, liver injury occurs by multiple mechanisms. Lipids cause lipotoxicity and lipotoxic stress in the endoplasmic reticulum and mitochondria, thereby causing apoptosis to occur.35 IRE1, PERk, and ATF6 converge at the C/EBP homologous protein to join with c-Jun to upregulate p53, a modulator of apoptosis, and express B cell lymphoma 2-associated X protein, which results in the release of cytochrome c.35,36 IRE1 also activates apoptosis signal regulating kinase 1 and c-Jun N terminal kinase (JNK) to form the c-Jun/C/EBP homologous protein complex, further promoting apoptosis and hepatic damage.37
Studies have shown that cholesterol crystals are present in the livers of human NASH and murine NASH models.38 KC can take up cholesterol-rich lipoproteins using scavenger receptors.39 Lipid droplet-laden KC recruit CD4+ and B lymphocytes.28 It has been shown that decreasing cholesterol levels causes dissolution of cholesterol crystals and disperse KC structures, helping to resolve NASH.40 Recently, it has been shown that cholesterol crystals activate NLRP3 in LPS-exposed KC and MCC950 small molecule inhibitor can inhibit the activation of NLRP3.41 Additionally, current research has shown that HepG2 cells exposed to low-density lipoprotein cholesterol formed cholesterol crystals on the lipid droplet membrane of hepatocytes and activated THP1 macrophage cells that upregulated TNF-α, NLRP3, and IL-1β mRNA (Figure 3).42
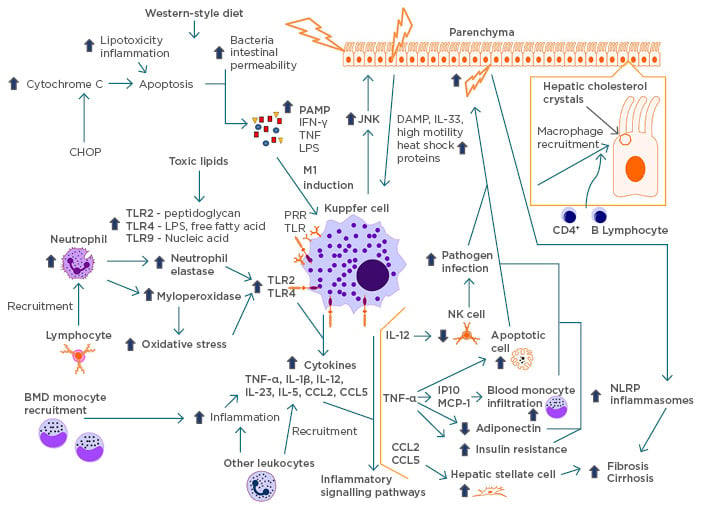
Figure 3: Cycle of inflammation.
A schematic modelling of the pathogenesis of NASH and its inflammatory cycle. Lifestyle factors, such as high-fructose diet and excess saturated fatty acids, lead to lipotoxicity, inflammation, bacterial, and intestinal permeability, causing damage to liver cells. These cells release PAMP, causing the induction of KC into the inflammatory M1 macrophage and recruitment of KC to the damaged sites by JNK. Active scavenging receptors recruit the macrophages with lipid droplets originating from hepatic cholesterol crystals and recruit other lymphocytes. The upregulation of TLR is due to the recruitment of neutrophils by activated lymphocytes and toxic lipids causing an increase in neutrophil elastase, myeloperoxidase, and oxidative stress. In response, KC release cytokines that initiate inflammatory pathways, thereby cascading the area into high inflammation. KC recruit BMD monocytes and cause more inflammation, releasing more cytokines. The inflammation leads to further release of DAMP in the hepatic cells, recruiting more KC. This cycle can eventually lead to fibrosis and cirrhosis.
BMD: bone mineral density; CHOP: C/EBP homologous protein; DAMP: damage-associated molecular patterns; JNK: c-Jun N terminal kinase; IFN: interferon; IL: interleukin; KC: Kupffer cells; LPS: lipopolysaccharide; NASH: nonalcoholic steatohepatitis; NK: natural killer; NLRP: NOD-like receptor protein; PAMP: pattern-associated molecular patterns; TLR: toll-like receptor; TNF: tumour necrosis factor.
Neutrophils have exhibited a role in KC activation and NASH progression through the attraction of lymphocytes and the release of myeloperoxidase that increases oxidative stress. In neutrophil-deleted mice, the activation of KC was delayed.43 Neutrophil elastase is thought to activate TLR2 and TLR4 receptors.43 Activation of KC and TLR4 has been demonstrated to co-ordinate neutrophil adhesion in liver sinusoids.44 Neutrophils recruit macrophages using an antigen presenting method. Additionally, neutrophil peptides or α-defensins have the capability to induce fibrosis by recruiting hepatic stellate cell proliferation.45 Aside from the role of KC in inflammation in NASH, KC also regulate metabolic activities and lipid metabolism of hepatocytes by expressing TNF and IL-1β.46 In NASH, there is a high uptake of lipids by KC via the secretion of lipases, lipid binding proteins, and bioactive lipids.
KC are activated by CD14, a PRR, when presented with LPS by the mediator TLR4. In cells with a high expression of CD14 there is an increase in sensitivity to LPS.47 Additionally, KC are activated on NF-κB, MAPK, ERK1, p38, JNK, and IRF3. In NAFLD, there is a high presentation of TLR4 expression resulting in a large release of cytokines, thus contributing to the pathogenesis of liver disease by furthering inflammation and causing fibrogenesis.
In the progression of worsening conditions from NAFLD to NASH, cells may release stress signals. During necrosis, cells release DAMP and chemo-attractants that can recruit various immune cells to the liver, initiating a wound healing response through fibroinflammatory repair. This can activate a full inflammatory response leading to fibrosis and cirrhosis (Figure 3). The cytokines produced by PRR lead to liver inflammation by the release of several cytokines and chemokines, such as TNF-α, IL-1β, IL-12, IL-23, IL-6, CCL2, and CCL5. The release of these cytokines results in the release of DAMP, promoting additional hepatocyte injury, activating TLR, JNK, and a cycle of vicious inflammation. Cytokines recruit leukocytes to further increase the inflammatory response in the healing process (Figure 3).
The recruitment of Ly-6C+ bone marrow derived monocytes via a CCR2-CCL2 recognition event has been shown to be a critical event for the promotion of steatohepatitis and NASH.48 Other chemokine interactions, such as CCL1–CCR8, CCL5–CCR1/CCR5, and CXC motif chemokine ligand 10 (CXCL10)-CXCR3, have been shown to recruit monocytes as well.49 These bone marrow derived monocytes can replace the resident KC, promote inflammation, and are identified as Ly-6Chi, CD11bhi, MHC IIneg, and CX3CR+.50 CXCL10 has been shown to enhance inflammation by inducing chemokines and cytokines such as TNF-α and IL-1β.51 Additionally, chemokine receptor CXCR3, a CXCL10 receptor, mediates inflammatory cytokines and macrophage infiltration (Figure 3).52
IL-12 expression leads to the loss of natural killer cells, resulting in a susceptibility to the increase of inflammation due to pathogen infections.53 CCL2 and CCL5 have overlapping properties that activate hepatic stellate cells leading to fibrosis. Additionally, JNK activation recruits macrophages to the site of hepatic inflammation, thus increasing inflammation and cell death.54
TNF-α has been shown to be a key component of NASH by promoting blood monocyte infiltration through the production of IP-10 and MCP-1 cytokines. TNF-α can activate proapoptotic or antiapoptotic signalling cascades, thereby controlling inflammation. TNF-α antagonises adiponectin, an anti-inflammatory adipocytokine, increasing inflammation, and also induces insulin resistance. The increase of TNF-α has been shown to have a crucial role in the development of NASH by promoting this inflammation.55
TREATMENTS
Categorisation
On 11th August 2017, there were 218 registered clinical trials under the search term ‘NASH’ on ClinicalTrials.gov. Macrophage-directed therapies to treat NAFLD and NASH promise to be a worthy intervention strategy. The many different roles and actions in the KC response system allows for different novel therapeutic approaches. These can be categorised into KC activation, KC polarisation, and monocyte recruitment.
Activation of Kupffer Cells
Preventing activation of KC by modulating TLR4 has been shown to ameliorate hepatic inflammation and injury. Transmembrane BAX inhibitor motif-containing 1 promotes the lysosomal degradation of TLR4 to inhibit insulin resistance, inflammation, and hepatic steatosis in mice and monkeys.56
M1 and M2 Polarisation
Carotenoids that inhibit lipid peroxidation exhibit antioxidant and anti-inflammatory effects in mice, in addition to regulating M1 and M2 activation, thus suggesting their important value for NASH treatment.57 Retinoic-acid-related orphan receptor α boosted M2 type in KC by activating Kruppel-like factor-4 and upregulating IL-10 in mice, leading to a reduction in inflammation and protection in NASH.58
Preventing the formation of cholesterol with cholesterol-lowering drugs and blocking TLR activation with ethyl pyruvate, phenylmethimazole, or other inhibitors both reduce and antagonise DAMP and PAMP in mice.40,59 Inhibiting the development of the inflammasome by inhibiting caspase 1, 8, and 9 with GS9450 has been shown in a Phase II trial to be a promising treatment option for patients with NASH.60
Blocking the inflammatory signal pathways of KC by inhibiting NF-κB, MAPK, ASK1, ERK1, p38, JNK, and IRF3 can also reduce inflammation in NASH. The ASK1 inhibitor, selonsertib, and the CCR2/CCR5 inhibitor, cenicriviroc, have both been shown to reduce fibrosis in mouse models and early clinical trials.37,49 Galectin-3 inhibitors have been shown to reduce fibrosis by inhibiting TGF-β mediated myofibroblast activation in mice.61 Andrographolide has been shown to inhibit NF-κB and NLRP3 inflammasome experimentally in mice.62
Monocyte Recruitment
Inhibiting inflammatory monocyte recruitment to the liver by interfering with the chemokine pathways CCL2–CCR2, CCL1–CCR8, CCL5–CCR1/CCR5, and CXCL10–CXCR3 have been shown to help in clinical trials with the CCR5 antagonist maraviroc and the CXCR4 antagonist plerixafor.49 Cenicriviroc blocks CCL2 recruitment of monocytes in addition to its antifibrotic effects.63 Reduction of TNF inflammatory cytokines with venlafaxine-103 has shown to reduce inflammation, steatosis, and cell death in alcoholic liver disease.64
Elafibranor (GTF-505), a PPAR α/δ modulator, has been shown in preclinical trials to decrease steatosis, inflammation, and display antifibrotic properties.65 GFT505 has also been shown to significantly improve steatohepatitis, fibrosis, and inflammation in humans through the regulation of PPAR.66 Honokiol exhibits an agonistic effect on PPARγ ligand-binding domains alleviating inflammation in mice.67 It was shown that the regulation of insulin, gluconeogenesis, glycogenolysis, and triglycerides through the targeting of farnesoid X receptor by bile acids provides anti-inflammatory and antifibrotic benefits. Obeticholic acid has been shown to resolve NASH through many pathways, including inhibiting hepatic lipid synthesis and inducing lipid uptake by adipocytes.68 Aramchol, a synthetic two component lipid molecule, has been shown to reduce hepatic fat levels considerably in animal studies.69 Additionally, treatment with neutrophil elastase inhibitor ameliorated glucose tolerance and steatosis in mouse models.70
Recently, an anti-inflammatory antibody-drug conjugate composed of the synthetic GC dexamethasone linked to an antibody for macrophage receptor CD163 exhibited a reducing effect of cytokines in experimental tests on rats, thus preventing steatohepatitis without apparent serious systemic side effects.71 With the shift from a two-hit hypothesis of NASH to a multivariable process, the therapeutic target of regulating the hepatic cholesterol metabolism became a key strategy in treating NASH. Regulating the SREBP2 and miR-33a genes with natural antioxidants suppresses triglyceride infiltration and fibrosis in cellular and murine models.72
CONCLUSION
It has become clear over the past decades that hepatic macrophages are central to initiating and propagating hepatic inflammation. Targeting these macrophages seems to be a promising therapeutic approach to treating NASH. Overall, there are many therapeutic options being discovered and tested to treat NASH by targeting macrophages. In the USA, >20% of patients with NAFLD progress to NASH; as many as 25 million adults in the USA have some form of NASH. These studies have many implications in the lives of patients with NASH.
There are still challenges that need to be overcome in targeting human liver macrophages. Firstly, although there is substantial similarity between mouse models and humans, there are many differences too. Experimental conditions vary and are not the same as human diseases. Secondly, many of the therapies alleviate fibrosis to some extent, but not fully resolve NASH. Therefore, although the clinical implications of alleviating fibrosis are very beneficial, the need to find a more encompassing treatment remains. Additionally, the cellular and molecular mechanisms of the progression to hepatocellular carcinoma and its significance in patients with NASH is a topic that still has much to be discovered.
There remains many points in the pathways of NASH that can be studied and explored for their therapeutic potentials. Targeting NASH in its early pathogenic stages may be a superior method compared to trying to reverse the damage done at later stages of NASH and cirrhosis.