Abstract
Haemochromatosis is an iron overload disorder that can be inherited or acquired and when diagnosis is delayed, disease progression and death can occur. Iron overload was first described by the French internist Armand Trousseau in 1865 in an article on diabetes in which alterations in skin pigmentations were reported. Some years later, the German pathologist Friedrich Daniel von Recklinghausen coined the term ‘haemochromatosis’ for a metabolic disorder characterised by excess deposition of iron in the tissue. This disorder affects 1 in 200 subjects of Caucasians of Northern European descent. The systemic excess iron build-up condition quickly gained an intense clinical interest. Haemochromatosis can lead to severe pathological symptoms in multiple organs, including the liver, bones, spleen, heart, pancreas, joints, and reproductive organs. With the progress of the disease, hepatic damage predominates. Polymorphisms in several independent genes can lead to haemochromatosis. However, the most widely known haemochromatosis-associated and studied ones are genetic variants in the HFE gene, located on the short arm of human chromosome 6. Early detection and phlebotomy prior to the onset of fibrosis/cirrhosis can reduce morbidity and normalise life expectancy. Consequently, phlebotomy has been accepted for decades as a standard treatment for the reduction of iron load. Nowadays, other methods, such as erythrocytapheresis, therapeutic application of iron chelators and proton pump inhibitors, or hepcidin-targeted therapy, are discussed as alternative personalised treatments of hereditary haemochromatosis. This review focusses on the pathogenesis, diagnosis, and therapy of haemochromatosis.
NORMAL METABOLISM OF IRON AND HEREDITARY HAEMOCHROMATOSIS
Iron is an essential micronutrient that exists in different ionic states. This ability makes it a critical component of oxygen transport proteins, numerous metabolic enzymes, and reaction centres of a large variety of redox enzymes. Most of the body’s iron is found in two proteins; around 70% of all iron is bound to haemoglobin in the red blood cells, while 15% is stored in myoglobin in muscle cells. The rest is stored as ferritin or haemosiderin in the liver, the spleen, and the bone marrow. Dietary iron is absorbed in the duodenum by enterocytes, in which Fe2+ is oxidised to Fe3+ in a ferroxidase reaction and transferred in the blood to transferrin. Subsequently, the iron-loaded transferrin binds to the transferrin receptor (TfR) and is taken up by receptor-mediated endocytosis (Figure 1A). In the cell, iron is released from this complex, while transferrin and its receptor are recycled to the cell membrane.
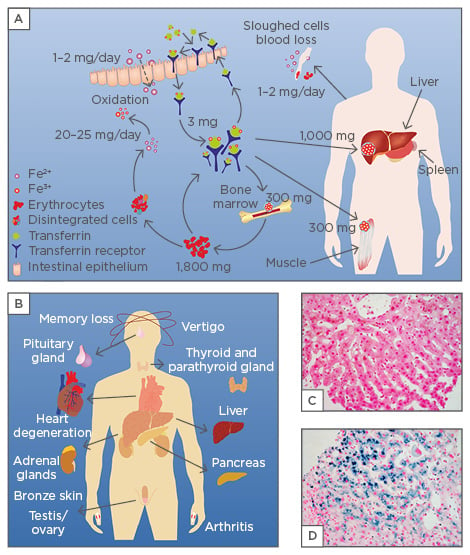
Figure 1: Body iron metabolism and iron overload.
A) Dietary iron is absorbed as part of a protein or in its ferrous Fe2+ form in the duodenum by enterocytes. In these cells, a ferroxidase reaction oxidises Fe2+ to Fe3+ and transfers it in the blood to transferrin, which has two specific high-affinity Fe3+ binding sites. Under physiologic conditions, there is approximately 3 mg iron bound to transferrin. The iron-loaded transferrin can bind to the transferrin receptor that is exposed to the cell surface and transported into the cell by receptor-mediated endocytosis. The main sites of iron storage are the liver (1,000 mg), muscles (300 mg), bone marrow (300 mg), and the erythrocytes (1,800 mg). Approximately 20–25 mg of iron per day is recycled in the reticuloendothelial system, which breaks down aged erythrocytes, while the total amount of iron through blood or sloughed cell loss is estimated at an average of 1–2 mg per day. B) Clinical manifestations of iron overload typically occur in the liver, pancreas, heart, adrenal glands, pituitary gland, and skin. Respective alterations provoke bronze skin pigmentation, hair loss, impotence, joint pain, lethargy, vertigo, and restriction of cognitive abilities.95-98 Elevated concentrations of iron in cardiomyocytes results in heart cell degeneration reflected by arrhythmias and cardiomyopathy. In the liver, the elevated concentration of iron causes hepatomegaly and liver cell necrosis that is indicated by elevated liver enzymes and formation of fibrosis and cirrhosis. C, D) Nuclear fast red/Prussian blue stain of a liver specimen obtained from a healthy subject (C) and a patient with hereditary HFE (D). The deep blue stain in D) is due to tissue iron deposits that are not detectable in healthy subjects.
Elevated iron quantities can be extremely toxic because of iron’s ability to donate and accept electrons, catalysing the conversion of hydrogen peroxide into free radicals, which can cause cell injury and organ damage.1,2 Haemochromatosis is characterised by excess endogenous iron stores in the body’s tissues and organs, particularly in the skin, spleen, heart, liver, pancreas, and joints (Figure 1B). Because humans themselves cannot increase the excretion of iron, untreated haemochromatosis can lead to bone and joint disease, cirrhosis, liver cancer, diabetes, hypothyroidism, hypogonadism, infertility, impotence, depression, or premature death due to liver or heart failure.3 The liver, as one of the main iron storage compartments and the place of hepcidin synthesis, is the central organ involved in maintaining iron homeostasis in the body. Overload of iron is a primary risk factor in the pathogenesis of liver disease.4,5 In affected patients, the condition of iron overload can be impressively demonstrated by the prominent stainable iron in liver sections by common histopathology stains using Prussian blue detecting iron deposits in biopsy specimens (Figure 1C and 1D). There is currently a debate on the impact of haemochromatosis on the development of retinal degenerative changes. While experimental studies performed in mice showed that retinal pigment epithelium from iron-overloaded mice exhibited a hyperproliferative phenotype, recent studies in patients with hereditary haemochromatosis (HH) revealed no indication of a higher prevalence of retinal degenerative changes.6,7
ABSORPTION AND IRON RECYCLING
The daily dietary iron absorbed by the body is strictly balanced. In industrialised countries, the body iron store contains 4–5 g, which is predominantly stored as haemoglobin in erythrocytes, and in different complexes in the liver, bone marrow, and muscles (Figure 1A). Typically, the daily iron loss due to sloughing of epithelial cells or blood loss is about 1–2 mg, while the normal reference range for serum iron is 65–176 μg/dL for men, 50–170 μg/dL for women, and 50–120 μg/dL for children. In healthy subjects, these iron levels predict a transferrin saturation (indicating the ratio of serum iron and total iron-binding capacity) of 20–50%.8,9 Red blood cell haemoglobin, liver, muscles, and macrophages of the mononuclear phagocytic system distribute the iron to locations in the body where iron is needed for proper cellular metabolism and respiration.10 The uptake capacity is regulated to a large extent by two factors: the size of the stored iron and the rhythm of erythropoiesis.11 The initial dietary iron is absorbed in the intestinal epithelium. There, the initial oxidised form of iron (Fe3+) is reduced by the apical ferric reductase duodenal cytochrome b that is expressed in the duodenum. The reduced iron (Fe2+) is then transported across the duodenal mucosa into the cytoplasm of the intestinal epithelium via the divalent metal transporter 1 (DMT1) by a proton-coupled mechanism.12-14 A part of the absorbed iron might remain bound on ferritin in the enterocytes, from where it will be removed from the body with shed enterocytes, thereby eliminating iron by faecal excretion.15 Iron transport from the enterocyte into the blood is mediated by ferroportin-1 (FPN1 or SLC40A1). This transmembrane protein is the only identified iron exporter from the basal membrane, macrophages, and hepatocytes.16,17 In this process, SLC40A1 exports iron across the basal membrane, which is then oxidised by hephaestin representing a copper-dependent ferroxidase most highly expressed in the small intestine but also in other organs.18,19
In the circulation, Fe3+ is bound to plasma transferrin representing the major iron storage source for the production of new erythrocytes in the bone marrow.20 It is utilised by erythroblasts (red blood cell precursors) for the synthesis of haemoglobin.21 Erythrocytes also participate with the macrophages in iron recycling. When aged erythrocytes are recognised by macrophages and erythrophagocytosis in the phagolysosome occurs, iron is recovered by the degradation of haemoglobin and either stored in ferritin or routed back to the circulation by the SLC40A1 transporter across the plasma membrane.22-24
TRANSPORT
Daily iron losses in healthy individuals are mainly due to bleeding (menstruation, pregnancy) and the regular exfoliation of iron-bound epithelial cells in the mucosal line of the gastrointestinal and urinary tracts, as well as from skin.25 There is no other effective way of iron removal, and existing iron has to be stored or used. A healthy individual typically contains 4–5 g of iron. More than half of this transition metal is bound in haemoglobin to transport oxygen, while the rest is bound in ferritin complexes. The liver represents the main store of iron and the hepatic stores of ferritin are the primary physiologic source of reserve iron in the body.26 Furthermore, it participates in absorption, storage, and export of iron, indicating that it is a key organ for iron regulation. The TfR system is the major mechanism for iron uptake in the liver and other tissues. Under low iron concentration levels, the expression of TfR1 is increased in the hepatocytes to promote cellular iron intake.27 Thus, the TfR primarily maintains cellular iron homeostasis. Conversely, at high iron concentrations, iron-regulating proteins are inactivated and TfR1 expression is suppressed, thereby lowering iron uptake. Studies in mice and humans have shown that subjects lacking transferrin develop massive iron overload in non-haematopoietic tissues, including liver and pancreas.28,29 Excess liver iron is known to promote steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma.30,31 If the iron concentration is too low, stored iron can be exported from the hepatocytes into the bloodstream by the transmembrane protein SLC40A1.16
REGULATION
The body’s iron concentration is very tightly regulated. The intracellular supply and storage of iron is mediated principally by three proteins: transferrin, TfR, and ferritin. Transferrin is a serum glycoprotein responsible for transporting bound iron from absorption centres to tissues.32 Furthermore, transferrin as a component of an iron-sensing system is crucial for the maintenance of iron homeostasis by modulating hepcidin expression.33 Most cells modulate iron uptake by regulating the amount of TfR expression and activity. Regulation is mediated by intracellular iron levels and iron-responsive elements within cell mRNA that are recognised by special iron regulatory proteins.32 The hepatic iron-regulatory hormone hepcidin, a major regulator of iron, is secreted into the blood and interacts with enterocytes to regulate the rate of iron absorption.
Hepatocytes have been proved to be the main source of the circulating hepcidin. Hepcidin is feedback regulated by iron concentration.34 When iron is abundant, more hepcidin is produced by hepatocytes limiting further iron absorption and release from iron stores. In case of low iron levels or deficits, lower production of hepcidin allows more iron to enter the circulation. It is believed that the liver uses transferrin saturation as a critical iron sensor, since the response of the liver-derived hormone hepcidin to dietary iron is induced by increased iron amount bound to plasma transferrin.35 There are two major cell iron uptake pathways: the transferrin-bound iron uptake and non-transferrin-bound iron uptake.36,37 Physiologically, the transferrin-bound iron uptake mechanism takes place. In this pathway, iron is internalised after binding on TfR1. After endocytosis, a ferrireductase reduces the released ferric iron and the reduced form is transported by DMT1 to the cytosol and the labile iron pool from which it can be mobilised when needed.37 The second, less characterised, non-transferrin-bound iron uptake mechanism is usually utilised in iron overload conditions when transferrin is saturated with iron. In this case, there is a necessity for the action of more cell surface ferrireductases or reductants (e.g. ascorbic acid) that will reduce ferric, non-transferrin bound, iron to its ferrous state, allowing its intracellular import by transporters such as DMT1.13,37,38
FIRST STEPS TO IDENTIFY HEREDITARY HAEMOCHROMATOSIS
The term haemochromatosis was coined in 1889 by the German pathologist von Recklinghausen who identified a patient with excess tissue iron deposits at autopsy.39 In 1935, the English gerontologist Joseph H. Sheldon suggested that this disease was an inherited metabolic disorder associated with increased iron absorption.40 Following these fundamental findings, it is now widely accepted that the iron overload is the outcome of increased intestinal iron absorption. The abnormal absorption by the gastrointestinal mucosa leads to excessive toxic quantities of iron in tissues that can provoke functional impairment of various organs, particularly in the heart and liver.41 In the mid 1950s, the first encouraging results in treating patients with therapeutic phlebotomy were reported.42 Since then, blood removal has been the method of choice for reducing excess quantities of iron. In 1975, HH was associated with the major histocompatibility complex (MHC) located on the short arm of human chromosome 6.43 However, the precise genetic basis for the development of HH was unravelled in 1996 when Feder et al.44 used linkage-disequilibrium and full haplotype analysis and identified a gene related to the MHC Class I family, initially termed human leukocyte antigen associated haemochromatosis (HLA-H), which was mutated in patients with HH.44
Nowadays, the HLA-H gene is referred to by its approved gene name HFE (high iron Fe-), which was given by the Gene Nomenclature Committee of the Human Genome Organisation (HUGO). It encodes a membrane protein critically involved in the regulation of circulating iron uptake. The HFE protein is primarily expressed in liver, intestinal, and, to a lesser extent, in immune cells.45,46 It interacts with the TfR required for iron import from transferrin into cells by endocytosis and is further critically involved in the regulation of the hepcidin gene (Hepcidin Antimicrobial Peptide [HAMP]), which reduces dietary iron absorption across the gut mucosa.
Studies on the HFE gene have revealed >20 mutations responsible for the majority of cases of Type 1 HH. However, the penetrance of these mutations is variable and a single mutation (i.e. Cys282Tyr or C282Y) in the HFE gene explains 80–90% of all diagnosed cases in populations of Northwestern European ancestry, while the penetrance of another variant (e.g. His63Asp or H63D) as well as their compound heterozygote forms (C282Y/H63D) are less frequently associated with the disease.47 HFE mutations lead to inadequate production of hepcidin, which results in increased iron uptake by the enterocytes, the release of iron from macrophages into the circulation, and increased deposition of excess iron in numerous tissues, which promotes excessive damage.48,49 HH and in particular the HFE-related type has been associated with liver damage, diabetes, insulin resistance, infertility problems, heart disease, stroke, neurodegenerative disorders, cancer, hormone imbalances, and artery disease.49-56
TYPES OF GENETIC HEREDITARY HAEMOCHROMATOSIS
A number of genetic alterations can result in the accumulation of excess iron and rise of haemochromatosis in the body. The most frequent are defects in genes encoding HFE, TfR2, ferroportin-1 (SLC40A1), HAMP, and hemojuvelin (HJV) (Figure 2A). All these defects are associated with elevated quantities of free intracellular Fe in the liver inducing formation of reactive oxygen species. These provoke oxidative damage of mitochondria, proteins, membrane structures, DNA, and lipids, thereby provoking cellular destruction. In the liver, the hepatocytes become necrotic and release large amounts of pro-fibrogenic cytokines, such as transforming growth factor-β, which induce inflammation, fibrosis, cirrhosis, and development of hepatocellular carcinoma.2
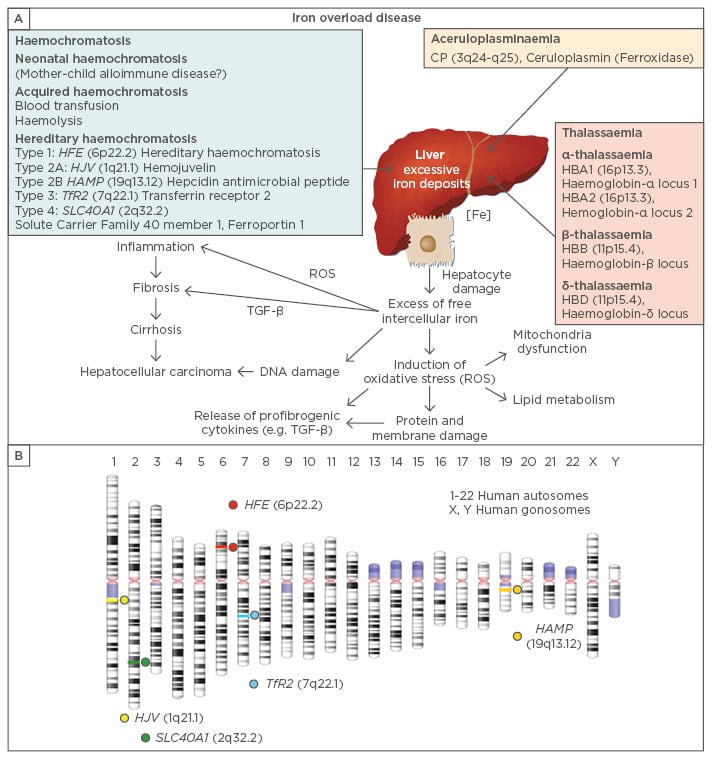
Figure 2: Genes involved in the pathogenesis of iron overload disease.
A) Excessive iron build-up may result from various mutations that impact iron homeostasis. Different types of hereditary haemochromatosis are induced by mutations affecting the genes HFE, HJV (hemojuvelin), HAMP (Hepcidin Antimicrobial Peptide), TfR2 (Transferrin Receptor 2), and SLC40A1 (Solute Carrier Family 40 member 1, Ferroportin 1). Other causes of Fe overload are neonatal haemochromatosis, acquired haemochromatosis, thalassaemia, or CP gene mutations encoding ceruloplasmin (also known as ferroxidase). In the liver, the elevated intracellular iron levels induce formation of reactive oxygen species (ROS), mitochondrial dysfunction, protein and membrane impairment, DNA damage, and lipid oxidation. These alterations lead to necrotic hepatocyte death and release of pro-fibrogenic cytokines, such as transforming growth factor-β (TGF-β), which induce inflammation, fibrosis, cirrhosis, and hepatocellular carcinoma.<sup>2</sup> B) Chromosomal localisation of the human ‘haemochromatosis genes’.
The different types of HH are classified by the underlying genetic mutations and mode of inheritance. Nowadays, five major genetic manifestations have been distinguished. Type 1 HH is by far the most intensively studied type and is linked to mutations in the HFE gene.44 The HFE protein associates with Class I light chain β2-microglobulin (β2m) and interacts with TfR on the cell surface, thereby reducing their affinity for iron and stimulating hepcidin expression.44,57,58 The Type 1 patients are most often homozygous for the C282Y mutated allele or carriers of compound heterozygote mutated alleles at amino acid position 282 and 63. In a cohort of >500 individuals, it was shown that homozygote C282Y carriers are genetically predisposed to accumulate iron and develop haemochromatosis with a prevalence of 34.3% for males and 29% for females to exhibit clinical symptoms.59 According to Seckington and Powell,60 the phenotypic spectrum of Type 1 haemochromatosis includes i) individuals with clinical HFE-HH who present organ damage as a result of harmful iron excess, ii) subjects with only biochemical HFE-HH where the iron accumulation is only reflected in increased transferrin-iron saturation and serum ferritin levels without impairment of organ physiology and function, and iii) non-expressing C282Y homozygotes who develop neither clinical symptoms related to HFE-HH nor iron overload.
Another much less common type of severe HH is juvenile Type 2 haemochromatosis, which in contrast to the Type 1 HH is a juvenile onset disorder. Juvenile forms of HH occur in two forms: Type 2A and 2B HH.61 Haemochromatosis Type 2A is also an autosomal recessive disorder that results from a mutation of the HJV (or HFE2) gene that is located on chromosome 1 (Figure 2B) and encodes the protein hemojuvelin which is produced in the liver, heart, and muscles.62 Hemojuvelin participates in the iron balance pathway by stimulating the expression of the iron regulating peptide hepcidin via the SMAD pathway.63-65 The individuals carrying this mutation have excess iron load already early in life, and develop symptoms in childhood. Respective individuals, by the age of 30 years, are characterised by hypogonadotropic hypogonadism, which leads to delayed puberty and infertility and risk for heart failure due to the prominent cardiac iron deposition.61 The juvenile haemochromatosis Type 2B is a phenotypically similar to Type 1 HH. It is caused by mutations within the hepcidin gene located on chromosome 19 and represents a key factor in the control of dietary iron absorption, storage, and tissue distribution.61,66
Type 3 HH develops typically before the age of 30 years, while Type 4 HH manifests similarly to Type 1 HH in adulthood, with clinical symptoms typically appearing between the ages of 40–60 years in men and usually after menopause in women. Type 3 is caused by mutations in the TfR2 gene, which is expressed on the basolateral surfaces of hepatocytes and a modulator of hepcidin expression in response to iron.66,67 Type 4 haemochromatosis, or ferroportin disease, is caused by mutations in the SLC40A1 gene encoding a cell membrane protein that has been suggested to be involved in iron export from duodenal epithelial cells and transfer of iron between maternal and fetal circulation.66 The symptoms of ferroportin disease vary from one person to another. The variety of symptoms is related to the different mutations of the SLC40A1 gene. Generally, ferroportin disease is divided into two forms: either a mild form or a more severe form resembling HH Type 1 in which liver damage and cirrhosis are more prevalent.68,69 Types 2, 3, and 4 are also mentioned in the literature as non-HFE HH types.66
HFE-MUTATIONS AFFECT IRON REGULATION
Type 1 HH is an autosomal recessive disorder resulting in iron overload and variable multi-organ dysfunction. HFE is a MHC class 1-like protein that associates with β2m enabling efficient transport of the HFE protein to the cell surface where it interacts with TfR1. It is thought that this protein functions to regulate circulating transferrin-bound iron uptake by regulating the interaction of the TfR1 with transferrin. The C282Y mutation disrupts the signalling bridges in the α-2 and α-3 extracellular domains of the HFE protein preventing the association of HFE with β2-microglobulin and TfR1, while the mutant protein is retained in the Golgi apparatus.70 The lack of the HFE/TfR1 interaction increases the affinity of TfR1 for the transferrin-bound iron, thereby modulating iron absorption by the duodenal crypt cells.71 In contrast to the C282Y mutation, the HFE-H63D variant maintains its affinity for TfR1, thereby supporting clinical findings showing that the H63D mutation only rarely leads to haemochromatosis. The modified molecular signalling between the HFE protein and the TfR1 is being intensively studied as the main regulatory mechanism in the pathogenesis of HH.72 Experimentation in mice deficient for the hepatic-HFE showed that iron homeostasis is predominantly controlled by regulating hepcidin that is produced in the liver and impacts the amount of iron absorbed from the diet and released from iron storage sites. Consistently, the over-expression of HFE is seen in mice lacking endogenous HFE upregulated hepcidin expression and normalised liver iron levels.73 In the same line, the lack of HFE in mice decreased the concentration of hepcidin, which strengthened further iron overload.74 Hepcidin expression can be modulated by mutations within HFE, HJV, and SLC40A1 that lead to iron disorders. Hepcidin is capable of regulating SLC40A1, affecting the iron absorption by enterocytes, iron export from macrophages into circulation, and deposition to cells or tissues.75
HFE DIAGNOSIS
Although practice guidelines for the diagnosis and management of haemochromatosis have been reported,76 new therapies are still in development with the aim to reduce discomfort and create quicker and permanent results. As discussed above, the majority of patients of Northern European descent with HH are homozygous for the C282Y mutation in the HFE gene. Although a significant proportion of patients with this genotype will develop iron overload (i.e. biochemical haemochromatosis), only a few will develop clinical symptoms and/or organ damage. The clinical penetrance depends on factors such as sex, age, genetic factors, and alcohol consumption.77,78 For the diagnosis of the disorder, the clinicians can rely on a combination of biochemical evidence of iron excess in the body, clinical symptoms, non-invasive testing, and imaging data. Suitable biochemical parameters that indicate iron overload are elevated serum transferrin-iron saturation values and serum ferritin concentrations. Although transferrin-iron saturation is an early and reliable indicator of risk for iron overload Type 1 HH, there is no correlation between transferrin-iron saturation and clinical findings. Likewise, untreated HFE-HH leads to increased levels of serum ferritin concentration.66 However, the serum ferritin level alone is not a specific marker of haemochromatosis because it belongs to the acute phase proteins that increase during inflammatory responses. Usually, elevated serum ferritin levels in association with elevated serum transferrin-iron saturation are more accurate for determining the disorder’s diagnosis.60 Additional testing could include a magnetic resonance imaging scan, which is a non-invasive method that allows measuring the degree of hepatic iron overload. If liver damage is suspected, quantitative iron measurements in a liver biopsy specimen might complete diagnosis.
The clinical symptoms indicating the progression of iron overload include hepatomegaly, hepatic cirrhosis, hepatocellular carcinoma, diabetes, cardiomyopathy, hypogonadism, arthritis, and progressive increase in skin pigmentation. Ultimately, genetic testing (C282Y, H63D, S65C) is suitable to confirm the diagnosis HH and to explain the occurrence of clinical symptoms and altered biochemical parameters. Subjects with abnormal iron tests should undergo genetic testing to confirm the diagnosis. In particular, several methods for genotyping of the C282Y, H63D, and S65C variant sites were established and are in daily routine use. These include restriction fragment length polymorphisms analysis, genomic sequencing, and real-time polymerase chain reaction analytics using fluorescence resonance energy transfer probes. The C282Y mutation disrupts a disulfide bridge that is located in the extracellular domain of the HFE protein, thereby preventing the association with β2m and the TfR and preventing the proper production of hepcidin.79 Since transferrin and hepcidin are both considered key elements in the regulation of iron absorption from the diet, iron regulation is disrupted and too much dietary iron is absorbed in the duodenum. This increase of dietary iron leads to the iron overload characteristic of Type 1 HH.73 The significance of the second most frequent HFE mutation, H63D, is still unclear.80 In addition, the HFE S65C mutation may lead to mild hepatic iron overload, but does not encounter for clinical manifest haemochromatosis in most patients.81 Likewise, a number of non-HFE mutations have been identified that are less commonly related to HH.
Aiming to identify individuals in a given population who are at higher risk for HFE-associated haemochromatosis, Eijkelkamp et al.82 developed a screening algorithm (Figure 3) using liver function test and transferrin saturation. Based on the general established guidelines, genetic testing is also recommended for all first-degree relatives of anyone diagnosed with HH, but general population screenings for HFE gene mutations remains a topic of debate.83-85 In the present guidelines, in patients with clinical suspicion and diagnosis of iron storage disease, iron imbalances are identified by increased ferritin and transferrin saturation (Figure 3). Genetic testing of relevant variant sites (C282Y, H63D, and S65C) is the gold standard to diagnose HH. If in respective patients, none of the variants occur, mutations in other genes that are involved in the control of iron homeostasis are most likely. Concomitant measurements of the hepatic iron index, which is the iron content given in µmol per g of liver corrected for the age of the subject, and scoring of existing fibrosis/cirrhosis, are further diagnostic options to estimate the success of therapy.86
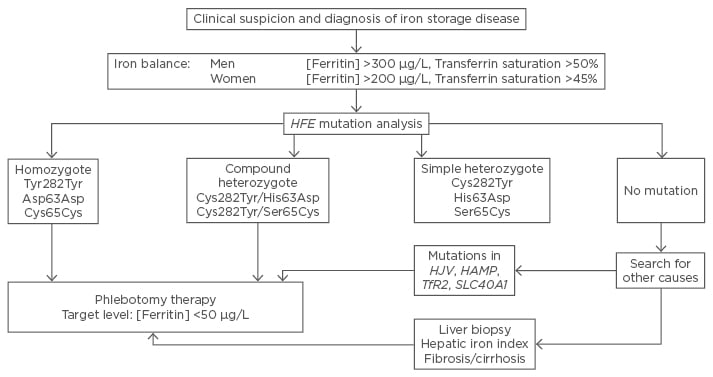
Figure 3: Diagnostic strategy in cases of suspected genetic haemochromatosis of Type 1.
A Type 1 HH can be diagnosed with certainty by detecting the mutations indicated. The detection of homozygous mutations in codons 282, 63, and 65, or the occurrence of a double heterozygous (‘compound’) mutation (Cys282Tyr/His63Asp or Cys282Tyr/Ser65Cys) is sufficient to diagnose HH. If no gene variants in the HFE gene are detectable, mutations in other genes (HJV, HAMP, TfR2, and SLC40A1) might be responsible for the iron overload condition. Therapy for iron overload in patients with proven mutations is done by regular bloodletting in which the target is a final ferritin blood level of <50 μg/L. The diagnostic strategy based on transferring saturation and genetic testing was first proposed by Eijkelkamp et al.82 HAMP: Hepcidin Antimicrobial Peptide; HH: hereditary haemochromatosis; HJV: hemojuvelin; SLC40A1: Solute Carrier Family 40 member 1.
The classical Type 1 HH is sometimes considered as a multifactorial disease characterised by mutations within the principal gene (i.e. HFE) and modulated by minor genetic or environmental factors that critically contribute to the outcome of the clinical phenotype in haemochromatosis. Nowadays, there is an increasing list of gene polymorphisms and factors known to contribute the penetrance of HFE gene mutations. However, the impact of these modifiers, including gene variants in HAMP, BMP2, FTL, and SLC40A1 genes, are not conclusive and are discussed controversially.87
THERAPY AND MANAGEMENT
HH is a disorder that causes an excess of iron deposition that can lead to toxic effects in tissues and multi-organ damage with clinical consequences that affect both quality of life and life expectancy. The aims in treating HH include diminishing of iron levels to the normal, preventing organ damage, treating possible complications such as liver disease, heart problems, or diabetes, and maintaining normal iron levels for the rest of life. Currently, the treatment of HH is based on phlebotomy therapy (venesection) and the elimination of iron by chelation therapy, depending on the aetiology.88 To date, phlebotomy is preferred, as it is more effective and more pleasant for the patient. In the initial phase, 500 mL blood is withdrawn, by the same procedure as blood donation, which leads to a decrease in body iron. After phlebotomy sessions, excess stored iron is utilised by the body to produce new red blood cells. During treatment, the quantities of remaining stored iron and the effectiveness of treatment are monitored by serum ferritin measurements. In addition, determination of haemoglobin levels helps to prevent a sharp iron decrease. The treatment should be done on a regular weekly basis, usually for 2 years. After reaching the desired iron decrease that is indicated by levels of serum ferritin, the patient undergoes (for the rest of their lives) maintenance phlebotomy every few months that is controlled by monitoring of transferrin saturation (ideally <50%) and serum ferritin (desired level 50–100 μg/L).89 Iron chelation is also a method to reduce iron pools in patients, particularly in those in which severe physical or medical difficulties in giving blood prevail. Therefore, this therapeutic regimen is to be classified as a more individualised treatment of HH. Iron chelating drugs presently used are: deferoxamine, which is administered by infusion, or deferiprone and deferasirox that are both given orally.88,90 While deferoxamine must be administered mostly on a daily base, the two oral iron chelators have several favourable effects in regard to toxicity, metabolisation, distribution volume, frequency of side effects, and compliance.90 These chelators successfully reduce hepatic and/or cardiac iron, but the application of these drugs is often associated with gastric discomfort, zinc depletion, musculoskeletal and joint pain, hearing impairment, visual disturbances, and skin irritations.90 Although the effective elimination of iron in individuals treated prior to the development of cirrhosis results in a normal life expectancy, subjects diagnosed after development of cirrhosis have decreased life spans even with iron depletion therapy.60 Also, nonhepatic manifestations, such as endocrine abnormalities and arthroplasty improve in only 20% of the patients that received iron depletion therapy.60
More recent findings suggest that the treatment with erythrocytapheresis is an alternative treatment. Erythrocytapheresis is the selective removal of erythrocytes by an extracorporeal apheresis procedure that leads to removal of more erythrocytes than during phlebotomy. As a consequence, this methodology requires drastically fewer treatment sessions necessary for the initial removal of overabundant iron.91 Several independent studies have proven that elevating hepcidin concentration is another possible strategy to ameliorate iron overload. Hepcidin-targeted therapies in which novel therapeutics manipulate the mechanisms regulating hepcidin production (e.g. hepcidin-based agonists, hepcidin expression, or activity stimulators) could provide a more fundamental future approach in HH treatment.34,89
In 2000, a clinical trial was initiated by the American National Institutes of Health (NIH) Clinical Center aiming to evaluate the effectiveness of a test using the mean corpuscular volume/haemoglobin monitoring guide for phlebotomy.92 This ongoing study compares the usefulness of the standard ferritin test in phlebotomy therapy in HH patients with that of mean corpuscular volume indicative for red blood cell size. Moreover, the trial will also clarify whether keeping iron levels low during maintenance therapy can improve liver disease and arthritis in affected patients.
Last but not least, major advances in molecular biology and pharmacology will lead to improving diagnosis and HH disease management. During any of the above-mentioned treatments, the reduction of iron overload is also managed by dietary changes. These include reduction in alcohol intake to prevent liver damage, avoiding iron containing supplements or drugs, as well as the restricted uptake of nutrients such as vitamin C that improves absorbance or dietary iron.
CONCLUSION
Since the first description of HH by the French internist Armand Trousseau in 1865, the understanding of the pathogenesis of this inherited disease and the regulation of dietary iron absorption has increased dramatically. In particular, genotyping and measurement of blood parameters indicating elevated concentrations of iron have enriched the diagnostic tools. During recent decades, special emphasis has been given to the understanding of iron metabolism and effective treatment of HH. The most effective treatment of HH is still the conduction of serial phlebotomies. Improving therapies and designing well-targeted iron chelating agents could minimise the toxicity, discomfort, and duration of current treatments, and lead to a higher quality of living standards in patients suffering from iron overload diseases. However, there are still challenges in haemochromatosis research that need to be addressed. Recent work has unravelled several gene mutations, polymorphisms, confounding factors, and environmental determinants impacting the outcome or severity of iron overload disease. The precise mode of activity of several of these modulating compounds is still enigmatic. More investigative studies aiming to decipher how these factors influence the clinical prevalence of iron overload are urgently needed. In addition, the complex network of iron-modulating and iron-regulating factors, such as hepcidin, will offer in the future a large variety of alternate promising therapeutic strategies for HH. In this regard, small cyclic agonists or peptides (i.e. the ‘minihepcidins’) of the iron regulatory hormone hepcidin that lower iron levels in the blood have already shown some highly beneficial effects in animals.93,94 In addition, the concomitant development of elementary guidelines for supervised application of reactive oxygen species scavengers would be helpful to prevent reactive oxygen species-associated cell damage and ongoing tissue damage in the setting of HH.