Abstract
Background: Hepatitis C virus (HCV) patients have a higher risk of developing renal impairment than health-matched controls. Fibrosis progression in HCV-related nephropathy could be accelerated. The role of angiopoietin 2 (Ang-2) in HCV-related nephropathy and its relationship with platelet parameters and thrombopoietin (TPO) is evaluated in this article.
Methods: Three patient groups were selected: HCV without nephropathy (n=90), HCV-related nephropathy (n=90), and controls (n=60). Laboratory analysis included complete blood count to reveal mean platelet volume and platelet distribution width (PDW), albumin creatinine excretion ratio, estimated glomerular filtration rate, and cryoglobulins. Quantitative real-time PCR, serum Ang-2, and TPO by ELISA, abdominal ultrasonography, and liver stiffness measurement by fibroscan were all conducted.
Results: Ang-2 was significantly higher in HCV-related nephropathy patients (43.0±36.9 pg/mL) when compared to healthy controls (16.6±4.3 pg/mL) (p=0.001). However, when compared to HCV without nephropathy (30.3±22.9 pg/mL), a statistically insignificant difference was noted (p=0.45). Logistic regression analysis revealed that significant fibrosis in HCV-related nephropathy is independently associated with platelet count (β: 0.98; p=0.000; odds ratio [OR]: 2.7), PDW (β: 0.722; p=0.000; OR: 2.1), serum TPO (β = 1.180; p=0.000; OR: 3.25), liver stiffness measurement by fibroscan (β: 1.29; p=0.000; OR: 3.63), and FIB4 (β: 1.07; p=0.000; OR: 2.9).
Conclusion: Ang-2, TPO, PDW, FIB4, and liver stiffness measurement are markers of liver fibrosis and portal hypertension in HCV-related nephropathy.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection represents a great health burden placing significant strain on healthcare resources. More than 180 million patients are chronically infected with HCV globally.1 Egypt has the highest seroprevalence of HCV, approaching 15% in 15–59-year-old patients in 2008; however, this has shown a promising decline after treatment was revolutionised with the introduction of direct-acting antiviral drugs.2
Patients with chronic active HCV have a greater possibility of developing renal impairment than their health-matched controls.3,4 The pathogenesis of hepatitis C-related nephropathy remains incompletely explained, but is mostly attributed to the deposition of HCV-associated immune complexes in the renal glomeruli and mesangial matrix causing renal injury.5
HCV-mediated renal injury could be explained by the presence of CD81 and SR-B1, which act as HCV receptors in the renal parenchyma facilitating viral entrance by endocytosis. HCV-induced upregulation of TLR3 induces mesangial proliferation and continuous immune pressure on B lymphocytes. Chronic HCV enhances the production of cryoglobulins, which are deposited in the mesangial matrix and glomerular capillaries, causing renal injury, glomerular fibrinoid necrosis, and thrombosis due to enhanced platelet aggregation.6 Insulin resistance induced by HCV enhances the production of intrarenal IGF-1 and TGF-β, which induces endothelin-1 expression, with reduced endogenous nitric oxide production perpetuating renal ischaemia.7
Angiogenesis can be stimulated under physiological conditions to allow hepatic regeneration, but when it exceeds these compensatory limits, such as in ischaemic hepatic conditions, it creates a hypoxic microenvironment, and thus angiogenesis had been previously labelled as one of the principle causes of disease progression in viral hepatitis.7,8 It is well known that angiogenesis is regulated and maintained by angiogenic cytokines, such as vascular permeability factor and angiopoietins.9
The angiopoietin/Tie2 signalling system represents an important regulator of angiogenesis. Angiopoietins, including angiopoietin 1 (Ang-1), bind to endothelial Tie2 and trigger autophosphorylation, which enhances endothelial cell survival, stabilisation, and vascular maturation, resulting in anti-inflammatory effects by blocking the effect of TNF-α on leukocyte migration. Tie2 activation induces trafficking of perivascular cells (vascular myocytes and pericytes) through paracrine endothelial substances. Ang-2 competes for Tie2 binding with Ang-1 and, as such, is considered an Ang-1 antagonist. The balance of Ang-1 and 2 is disrupted in chronic inflammatory states and neoplastic conditions, which enhances the vascular instability and leakage.10-12 Vascular permeability factor mediates the cellular effects of angiopoietins.13 Integrins, mainly integrin α5β1, have a role in upregulating Ang-1/Tie2 crosstalk.14
HCV structural proteins, mainly NS3, may have an important role in stimulation of proangiogenic factors and cytokines but the mechanisms underlying hepatic angiogenesis in chronic HCV-infected patients remain unclear and need to be evaluated.15
Thrombopoietin (TPO) is the principal regulator of megakaryocytopoiesis and thrombopoiesis and is mainly produced in the liver. Binding of TPO to its receptors expressed on the surface of the effector cells stimulates thrombopoiesis through enhancing the JAK/STAT signalling pathways. TPO levels are directly proportional to platelet count and are reduced in liver cirrhosis due to decreased production or due to a direct effect by hepatotropic viruses such as HCV.16 The current study aimed to assess potential markers of fibrosis progression in HCV-related nephropathy.
STUDY DESIGN
Patient Selection
This prospective case-control study was conducted in the outpatient clinic of the Internal Medicine and Hepatology Departments, Zagazig University Hospital, Zagazig University, Zagazig, Egypt, in the period from January 2014–March 2017.
Out of 1,435 patients with chronic active HCV who were evaluated for treatment with direct antiviral agents, 180 patients (12.5%) were enrolled in this study. Questionnaires regarding medical history, drug history, and family history of all participants were obtained. The inclusion criteria the study used were patients aged 18–60 years, with seropositivity for HCV antibodies, positive HCV RNA, and HCV-related nephropathy. The patients were classified into two groups: Group 1 included 90 patients with chronic active hepatitis C virus without nephropathy with a mean age of 45.2±6.2 years and Group 2 included 90 patients with chronic hepatitis C virus-related nephropathy with a mean age of 44.8±6.6 years. A control group included 60 healthy subjects matched for age and sex after exclusion of HCV, HBV, diabetes, hypertension, and nephropathy with a mean age of 43.9±4.5 years.
Exclusion Criteria
Exclusion criteria used to select patients for the study included causes of nephropathy other than HCV or any condition which may alter ANG-2 levels, such as patients with hepatorenal syndrome; hepatic decompensation; hepatitis B virus; diabetes; autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis; obesity or essential hypertension; current treatments that may induce nephropathy, such as non-steroidal anti-inflammatory drugs, D-penicillamine, gold, alcohol, and contrast dye; previous treatment with antiviral or immunosuppressive drugs; current urinary tract infections; malignancy; and pregnancy.
METHODS
All patients had a clinical examination to assess clinical signs of portal hypertension, such as dilated abdominal veins, splenomegaly, and the condition of the liver, whether shrunken or enlarged; exclusion of features of liver cell failure, such as jaundice, ascites, lower limb oedema, fetor hepaticus, flapping tremors, spider angiomas, palmer erythema; and clinical signs of nephropathy, such as oedema of lower limbs, haematuria, oliguria, hypertension, and signs of volume overload.
All procedures were performed in accordance with the ethical standards of the Zagazig University’s Faculty of Medicine research committee and with the 1975 Declaration of Helsinki and its later amendments. Written informed consent was obtained from patients for interview, anthropometric measurements, and blood sampling.
Laboratory Analysis
Complete blood count assessments were performed, including mean platelet volume (MPV) and platelet distribution width (PDW). Liver function tests were also executed, including assessment of total and direct serum bilirubin, serum albumin, serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), prothrombin time, prothrombin concentration (%), and international normalised ratio. Kidney function tests included blood urea and serum creatinine.
Albumin-creatinine excretion ratio was classed as normal up to 20 mg/g creatinine, and urine microalbuminuria was classed as normal up to 30 mg/24 hours. Estimated glomerular filtration rate (GFR) was calculated as 140–age x weight x 0.85 if female/72 x serum creatinine.17 The stages of GFR were classed as:
- Stage 1: 90 mL/min
- Stage 2: 60–89 mL/min
- Stage 3a and 3b: 30–59 mL/min
- Stage 4: 15–29 mL/min
- Stage 5: <15 mL/min or on dialysis
Estimation of cryoglobulins, rheumatoid factor, and complement levels in blood and fasting blood sugar were classified according to the American Diabetes Association (ADA) criteria 2010.
The HBsAg surface antigen, serum α fetoprotein, and HCV antibodies were detected using Cobas® e 411 (Roche Diagnostics GmbH, Mannheim, Germany), a third-generation, commercially available enzyme-linked immunosorbent assay kit.
Quantitative measurement of HCV load in patient sera was carried out by real-time, quantitative PCR (COBAS® AmpliPrep/COBAS® TaqMan® HCV Test, v2.0, Roche Diagnostic Systems Inc., Welwyn Garden City, UK), with a detection limit of 15 IU/mL.
Serum level of Ang-2 was measured by ELISA, following the manufacturer’s recommendations (Quantikine, R&D Systems, Minneapolis, Minnesota, USA). All measurements were made in duplicate. Absorbance was read at 450 nm and corrected at 570 nm. Assay range: 1.6–140.0 pg/mL.
Serum TPO level was measured using a commercial quantitative sandwich enzyme immunoassay (Quantikine Immunoassay Control Set 934 Human TPO, R&D Systems) with a reference range of 31–2,000 pg/mL.
Abdominal Ultrasonography
All patients were examined using a real-time grey-scale device by a transducer with a frequency of 2.5–5.0 MHz. The patients were examined after a 6-hour fast. Criteria for cirrhosis diagnosis were determined by a coarse, nodular appearance and shrunken size with prominent caudate lobe. Criteria for portal hypertension diagnosis included portal vein diameter >13 mm measured at point of crossing inferior vena cava, splenic bipolar diameter >130 mm, and splenic vein diameter 10 mm. Features of chronic renal disease were excluded.
Liver Stiffness Measurement
Liver stiffness measurement (LSM) was performed by a fibroscan. The number of shots was 10 and the interquartile range ≤25%. Generally, a liver stiffness of 2.5–7.0 kPa denotes F0–1, 7–9.5 kPa indicates F2, 9.5–12.5 kPa indicates F3, and >12.5 kPa denotes cirrhosis.18
Statistical Analysis
All data were collected, tabulated, and statistically analysed using SPSS 20.0 for Windows (SPSS Inc., Chicago, Illinois, USA). Quantitative data were expressed as the mean±standard deviation and median (range), and qualitative data were expressed as absolute frequencies, both numbers and percentages. For independent samples a t-test was used to compare between two groups of normally distributed variables while the Mann–Whitney U test was used for non-normally distributed variables. The F-test was used to compare more than two groups of normally distributed variables. The Kruskal–Wallis test was used to compare between more than two groups of non-normally distributed variables. Percent of categorical variables were compared using the chi-squared test or Fisher’s exact test when appropriate. The Pearson correlation coefficient was calculated to assess the relationships between various study variables. All tests were two-sided. A p value <0.05 was considered statistically significant. Logistic regression analysis was used to elucidate the independent relationships associated with significant fibrosis in HCV-related nephropathy and odds ratios were calculated from exponential beta.
RESULTS
The baseline laboratory, metabolic, LSM by fibroscan values of patient subgroups are shown in Table 1. HCV genotyping revealed that genotype 4a (n=94, 52%) and 4c (n=67, 37%) were predominant, mixed 4.1b (n=19, 11%). The mean values of serum TPO, platelet count, and PDW were significantly lower and aspartate aminotransferase/alanine aminotransferase and FIB4 were significantly higher in patients with nephropathy.
The mean value of LSM by fibroscan was significantly higher in HCV-related nephropathy patients (9.9±3.2 kPa) compared with HCV patients without nephropathy (7.0±1.6 kPa) (p=0.04). F4 or cirrhosis by fibroscan was diagnosed in 10 patients in the HCV-related nephropathy group (11%) versus no patients in the HCV without nephropathy group, which was statistically highly significant (p=0.007), as shown in Table 1.
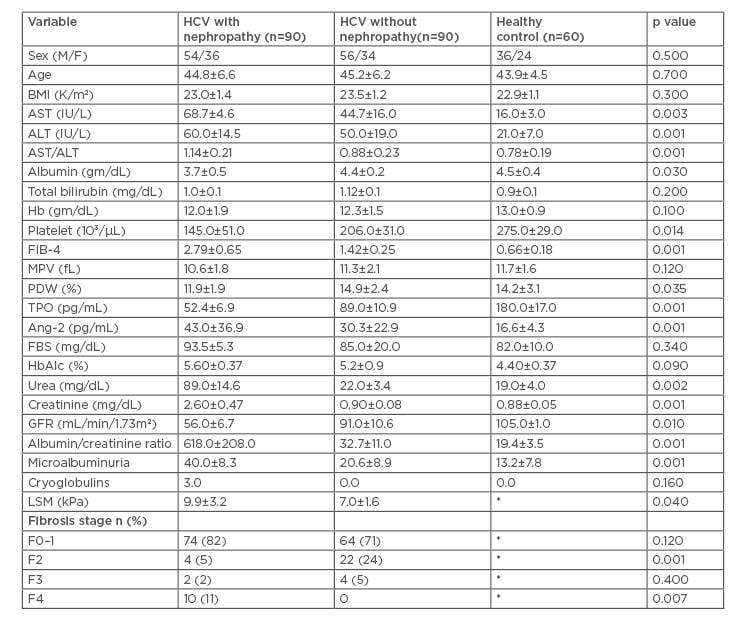
Table 1: Baseline laboratory, metabolic, and fibroscan values of patient subgroups.
*These tests were not performed for healthy controls.
ALT: alanine aminotransferase; Ang-2: angiopoeitin; AST: aspartate aminotransferase; BMI: body mass index; FBS: fetal bovine serum; GFR: glomerular filtration rate; Hb: haemoglobin; HCV: hepatitis C virus; LSM: liver stiffness measurement; MPV: mean platelet volume; PDW: platelet distribution width; TPO: thrombopoietin.
In the HCV nephropathy group, 48 patients (53.3%) (F4=10, F3=2, F2=4, F0–1=32) had higher values of Ang-2 (74.9±52.6 pg/mL) when compared to 16 patients (17.8%) in HCV without nephropathy (62.3±32.5 pg/mL) (F4=0, F3=4, F2=8, F0–1=4) and no patient in the healthy group showed a high value of Ang-2 (p=0.001). The mean value of Ang-2 was significantly higher in HCV related nephropathy group when compared to the healthy controls (43.0±36.9 pg/mL, 16.6±4.3 pg/mL, respectively; p=0.001). However, when compared to the HCV without nephropathy group (30.3±22.9 pg/mL), a statistically insignificant difference was noted (p=0.45).
In patients with nephropathy and thrombocytopenia, Ang-2 showed a significant negative correlation with platelet count (r=-0.780; p=0.001), PDW (r=-0.540; p=0.001), serum TPO (r=-0.802; p=0.000) (Figure 1), and GFR (r=-0.770; p=0.000), and a significant positive correlation with LSM by fibroscan (r=0.910; p=0.000) (Figure 1), FIB4 (r=0.823; p=0.000), creatinine (r=0.495; p=0.001), and microalbuminuria (r=0.418; p=0.004).
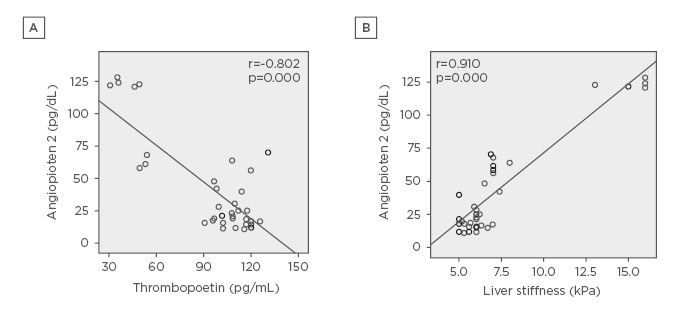
Figure 1: A) Negative correlation between thrombopoietin and angiopoietin 2 levels in HCV positive patients with nephropathy. B) Positive correlation between liver stiffness by fibroscan value and angiopoietin 2 levels in HCV positive patients with nephropathy.
HCV: hepatitis C virus.
Logistic regression was performed to determine variables independently associated with significant fibrosis in HCV-related nephropathy. The variables identified were platelet count (β: 0.98; p=0.000; odds ratio [OR]: 2.70), PDW (β: 0.722; p=0.000; OR: 2.10), serum TPO (β: 1.180; p=0.000; OR: 3.25), LSM by fibroscan (β: 1.29; p=0.000; OR: 3.63), and FIB4 (β: 1.07; p=0.000; OR: 2.90).
When the patients were stratified according to stages of fibrosis by fibroscan, Ang-2 showed a highly significant increase with progression of fibrosis stage compared to serum TPO, platelet count, MPV, and PDW, which showed a significant decrease (Table 2).
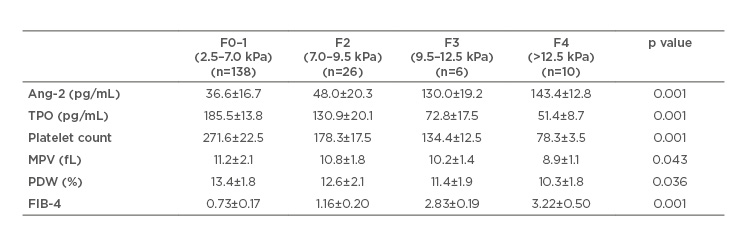
Table 2: Relation between fibrosis stage progression by fibroscan, angiopoietin 2, thrombopoietin, and platelet parameters in the studied patients.
Ang-2: angiopoietin; MPV: mean platelet volume; PDW: platelet distribution width; TPO: thrombopoietin.
DISCUSSION
Renal affection due to chronic hepatitis C is mainly due to immune-complexes or cryoglobulins deposition, vasculitic affection of the renal blood vessels, and a direct viral cytopathic injury.19 Subclinical renal involvement should also be highlighted due to the possibility of an underlying occult HCV infection.20 Still, these mechanisms of renal injury cannot explain all the documented lesions. In addition, the microscopic characteristics of HCV nephropathy have unique features which prove that the virus may induce renal injury via specific mechanisms, including induction of apoptosis, modulating caspases, and NS3 binding to TLR3, which induces mesangial proliferation.21
Vascular permeability factor, Ang-2, and matrix metalloproteinase 9 are concomitantly increased in chronic HCV and play an important role in vascular remodelling and fibrosis progression through perpetuation of inflammation, release of fibrogenic molecules from the stimulated endothelial cells, and directly affect hepatic stellate cells. This effect was supported by the increased expression of Ang-2 mRNA in liver biopsies taken from the study patients.22
A positive, significant correlation between Ang-2 and stage of hepatic fibrosis in chronic HCV patients would help in the diagnosis and monitoring for disease progression. A non-invasive model has been suggested that includes evaluation of platelets count, transaminases, and Ang-2.23 Serum values of Ang-2 and Ang-1 correlated with fibrosis progression in HCV patients. The ratio of Ang-2 and Ang-1 may prove to be a useful index for monitoring the progression of chronic liver disease.24
Previous research could not define a direct relation between portal pressure and platelet count; however, thrombocytopenia that persists after splenectomy may be corrected after successful liver transplantation.25
The level of TPO as the main factor affecting the level of circulating platelets remains variable in cirrhosis due to increased levels as a result of conditions associated with platelet destruction due to enlarged spleen or reduced levels in patients with advanced liver diseases.26 MPV is directly proportional to the rate of TPO production, therefore it is increased in cases of increased destruction. PDW can indicate the degree of difference in platelet size; however, their variability and use in liver cirrhosis is rarely discussed or studied. Increased MPV and PDW in cirrhosis denotes an increased destruction of platelets along with increased levels of young platelets in the blood if serum TPO is increased.
The current study showed a significant negative correlation between Ang-2 and serum level TPO, and platelet count, MPV, and PDW, as well as a positive correlation between FIB-4 and LSM by fibroscan. Platelet parameters and TPO were decreased in patients with advanced fibrosis or cirrhosis due to the occurrence of portal hypertension. This observation is supported by a study that revealed Ang-2 levels were elevated in patients with cirrhosis and hepatocellular carcinoma compared to the healthy controls (p=0.001) and correlated inversely with markers of synthetic liver function, such as serum albumin and prothrombin concentration, and correlated positively with markers of excretory function, such as serum bilirubin.27,28
There was a significant elevation of serum level of Ang-2 in patients with HCV infection when compared to the healthy controls, but insignificant difference was detected among HCV patients when stratified by the presence of nephropathy (p=0.45).
A significant positive correlation was found between Ang-2 and markers of renal injury in HCV as microalbuminuria (p=0.004). These findings were also supported by a study that showed that hepatitis C infection was independently associated with microalbuminuria in subjects without diabetes (OR: 1.99; 95% confidence interval: 1.38–2.85; p=0.008).29
This study did not investigate the impact of HCV eradication by direct-acting antiviral agents on the levels of Ang-2 or TPO. Patients with HCV related nephropathy were treated with ritonavir, ombitasvir, and paritaprevir, which was a limitation of the current study.
CONCLUSION
In conclusion, in HCV-related nephropathy, histological progression is anticipated. This can be predicted by platelet count, PDW, FIB-4, serum TPO, and serum angiopoietin 2, which may justify their use as markers of liver fibrosis and portal hypertension in patients with chronic HCV-related nephropathy.