Abstract
Primary sclerosing cholangitis (PSC) is an autoimmune chronic liver disease that studies have shown is rare in children. Here, a challenging case of PSC in a 13-year-old male, without preceding manifestations of inflammatory bowel disease and with evidence of biliary obstruction, is reported. The patient presented with progressive scleral icterus; their total bilirubin and alkaline phosphatase levels were raised, with negative autoimmune work-up, and an ultrasound scan of their abdomen was unremarkable. Magnetic resonance cholangiopancreatography revealed marked dilatation of the intra- and extrahepatic bile ducts, with strictures in the hepatic duct and proximal common bile duct (CBD). Endoscopic retrograde cholangiopancreatography revealed a very narrow CBD with high-grade severe biliary stricture at the common hepatic duct. A cholangiogram revealed the beaded appearance of the intrahepatic ducts, and a left percutaneous external biliary drainage tube was placed. Overall, the findings were suggestive of PSC with high-grade CBD strictures. This case is unique due to the absence of preceding clinical manifestations of inflammatory bowel disease and predominantly obstructive symptoms at the time of presentation, which is highly unusual in the paediatric population.
Key Points
1. Primary sclerosing cholangitis (PSC) usually presents in young and middle-aged males and is rare in children.2. Paediatric patients with PSC usually experience an insidious disease course, and initially present with inflammatory bowel
3. PSC is one of the widest unmet needs in hepatology, and although some therapies show promise, trials are yet to agree on an effective treatment for the disease.
INTRODUCTION
Primary sclerosing cholangitis (PSC) is an autoimmune chronic liver disease that is mostly prevalent in young and middle-aged males, less common in females, and rare in children. Morbidity and mortality rates are high for PSC due to its progression to end-stage liver disease (ESLD), which often requires a liver transplant. Epidemiology of PSC is not well-defined in the paediatric population: studies have shown that it is an extremely rare disease in children, with a reported incidence rate that is 20% less than that of adults.1 Most paediatric patients initially present with inflammatory bowel disease (IBD) and are found to have underlying PSC coincidentally. Unlike the adult population, PSC in paediatrics have an insidious course, with <5% of the paediatric population having dominant biliary strictures (DBS) or ESLD at the time of their diagnosis.2 Here, an unusual and challenging case of PSC in a 13-year-old male, without preceding manifestations of IBD and with evidence of biliary obstruction at the time of diagnosis, is presented.
CASE PRESENTATION
A 13-year-old male with no significant past medical history presented to the clinic with progressive yellow discoloration of the eyes for 3 weeks, associated with multiple episodes of non-bilious, non-bloody vomiting; fever; intermittent, generalised body itching; and significant weight loss. Their initial vital signs showed a blood pressure of 93/65 mmHg, heart rate of 112 beats per min, respiratory rate of 16 breaths per min, body temperature of 37 °C, and a pulse oximetry that was 98% on room air. Physical examination was significant for scleral icterus and a non-tender, non-distended abdomen with normal bowel sounds.
Initial lab work-up showed white blood cells: 6.70×103 /µL; red blood cells: 5.07×106 /µL; haemoglobin: 14.10 mg/dL; haematocrit: 43.3%; platelets: 441×103 /µL; total bilirubin: 3.80 mg/dL; direct bilirubin: 1.91 mg/dL; gamma-glutamyl transpeptidase (GGT): 96 U/L; alanine aminotransferase: 66 U/L; aspartate transaminase: 57 U/L; and alkaline phosphatase (ALP): 196 U/L. Further laboratory tests were negative for the hepatitis A antibody IgM, the hepatitis B core antigen IgM, and the hepatitis B surface antigen, and found that the level of liver–kidney microsomal antibody was <1.0 U, mitochondrial M2 antibody was <20.0 U, and smooth muscle antibody was 6.0 U.
An abdominal ultrasound revealed gallbladder wall thickening, with no shadowing calculus or pericholecystic fluid and with no common bile duct dilation. Magnetic resonance cholangiopancreatography revealed marked dilatation of the intra- and extrahepatic biliary duct, as well as areas of strictures seen in the hepatic duct and the proximal common bile duct (CBD). There was no evidence of pericholecystic fluid or a filling defect, which would suggest cholelithiasis or choledocholithiasis.
Endoscopic retrograde cholangiopancreato-graphy (Figure 1) revealed a very narrow CBD with a high-grade severe biliary stricture in the common hepatic duct. A cholangiogram revealed areas with a beaded appearance in the intrahepatic ducts. A papillotomy was performed, but the procedure was terminated as the guidewire could not bypass the severe obstruction of the biliary stricture in the common hepatic duct. A percutaneous transhepatic cholangiogram was performed by an interventional radiologist, which demonstrated a high-grade stricture in the mid CBD with pre-stenotic dilatation. A left percutaneous external biliary drainage tube was placed, resulting in an improvement in total bilirubin and ALP levels. An ultrasound-guided liver biopsy revealed liver parenchyma with focal and loose concentric fibrosis around the bile ducts and focal periportal fibrosis, confirmed by trichrome stain.
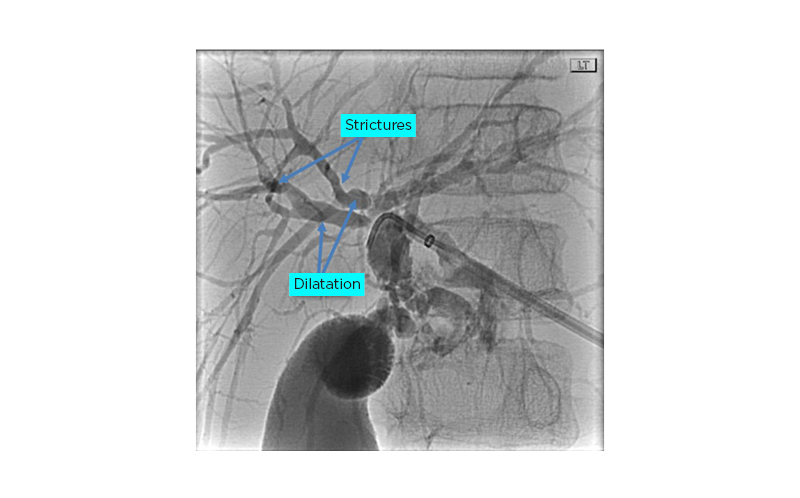
Figure 1: Fluoroscopy image demonstrates dilatation of the intra- and extrahepatic biliary ductal system.
Portions of the hepatic duct and proximal common bile duct demonstrate areas of stricture.
Overall, the findings were suggestive of PSC with high-grade CBD stricture. The patient was started on the fat-soluble vitamins A, D, E, and K, and ursodiol (13 mg/kg daily). Their faecal occult blood test was positive, and an oesophago-gastro-duodenoscopy and colonoscopy were performed since PSC is mostly associated with IBD, specifically ulcerative colitis (UC). An endoscopy showed normal oesophageal, gastric, and duodenal mucosa; a colonoscopy (Figure 2) with biopsy revealed colonic mucosa with chronic focally active colitis with crypt distortion in both the ascending and descending colon, confirming a diagnosis of UC. The patient was started on delayed-release oral mesalamine (4 g/day).
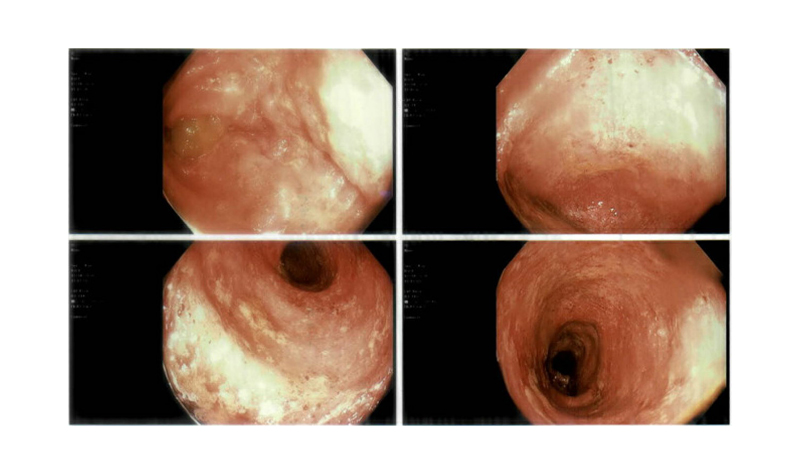
Figure 2: Colonoscopy images revealing erythematous and ulcerated colonic mucosa.
DISCUSSION
PSC in paediatrics has an insidious course, mild symptoms, and most cases initially present without complications. The most reported symptoms are fatigue, itching, weight loss, and right upper quadrant pain. Jaundice and obstructive biliary symptoms are highly unusual at the time of the patient’s presentation.2-4 Less than 5% of the paediatric population have DBS or ESLD at the time of diagnosis. The natural course is usually progressive, with 50% of children developing clinical complications and 30% requiring liver transplantation (LT) within 10 years from the time of diagnosis.5
A review of the literature indicated that 60–80% of cases of patients with PSC have associated IBD, an incidence that is thought to be higher in the paediatric population. The most common phenotype of IBD is UC, and at least 10% of children with UC are affected with PSC. Diagnosis of IBD usually precedes PSC by many years. Children with PSC-IBD often have more severe mucosal inflammation compared to those who have IBD without PSC.2,5 This case is unique due to the absence of preceding clinical manifestations of IBD and predominantly obstructive symptoms at the time of presentation, which is highly unusual in the paediatric population. This highlights the importance of considering PSC in children with features of cholestasis or abnormal liver function tests, even in the absence of IBD manifestations.
Most children with PSC have elevated levels of GGT and ALP early in the course of disease, but GGT is more specific in the paediatric population.1,3 The gold standard imaging is magnetic resonance cholangiopancreatography, with 89% sensitivity in children,6 and typical findings from this are dilated intra- and extrahepatic biliary ducts, with multiple areas of narrowing. The presence of dominant strictures of extrahepatic biliary tree is unusual at the time of diagnosis. As seen in this patient, DBS are most often found at the bifurcation of the hepatic duct.7,8 In the presence of typical cholangiography changes and negative autoimmune serologic markers, a liver biopsy is not typically required for the diagnosis of PSC.9
PSC is recognised as having one of the largest unmet needs in hepatology, as currently there is no proven medical therapy available to delay the progression of liver disease or the onset of complications.10 Management is mainly supportive and geared toward treating complications and palliating symptoms. The normalisation of ALP levels has been associated with improved prognosis,11 and LT has been proven to prolong survival in patients with PSC.12
Transhepatic and endoscopic balloon dilatation of strictures has been shown to be useful in the palliation of symptoms. Surgical drainage procedures (e.g., portoenterostomy and choledochoenterostomy) are associated with an increased risk of cholangitis and could make the subsequent liver transplantation technically challenging.8,13
There have been conflicting results regarding the role of ursodeoxycholic acid (UDCA). Multiple paediatric studies have shown that UDCA is more effective than a placebo in lowering serum ALP, serum bilirubin, symptoms of pruritis, and potentially the incidence of cholangiocarcinoma, but it consistently showed no survival benefit.8 A double-blind randomised controlled trial in adults assessing the efficacy of high-dose UDCA further complicated the use of this therapy, as significantly higher rates of death, LT, and other serious adverse events were seen in the drug-treated group, despite biochemical improvement observed.14 The current consensus is to avoid UDCA at doses >20 mg/kg/day, and that it could be beneficial in a subset of patients with PSC who tolerate it well and have normalisation of ALP.3,10
The gut microbiome and PSC dysbiosis (reduced microbiota diversity) has been implicated in the pathogenesis of the disease.15 Oral vancomycin therapy (OVT) has shown very promising results in two previous pilot studies in the paediatric population, as 6 weeks of OVT induced sustained remission in 14 patients with non-cirrhotic PSC, resulting in the improvement of symptoms and a reduction in biochemical markers, although there was no consistent improvement in liver histology.16,17 Similar effects in the reduction of liver biochemistry were reported in small-scale prospective adult studies. On the contrary, the largest retrospective study to date in paediatric patients by Deneau et al.18 showed that neither patients treated with OVT nor those treated with UDCA had outcome benefits compared to the non-treatment group, and the rate of progression to ESLD was similar between the clinical groups.18
PSC has a waxing and waning course of inflammation. During the early stages of disease, the spontaneous normalisation of biochemical markers is common in children.11 This characteristic feature in the natural history of PSC is likely to have contributed to the conflicting data and challenges in inferring results from the predominantly small and underpowered studies that are currently available in the literature.
CONCLUSION
Specifically in the paediatric group, PSC is a rare disease with poor prognosis. Major concerns for patients with PSC include the progression to ESLD and an increased risk of developing malignancies such as cholangiocarcinoma, gallbladder cancer, and colorectal cancer.13 Further prospective and long-term follow-up studies are necessary to expand the current literature in order to better understand and manage this disease.