Interview Summary
Progressive familial intrahepatic cholestasis (PFIC) is a cholestatic condition that typically arises in childhood. As such, if an adult presents with PFIC symptoms, diagnosis may be less clear-cut, or symptoms may be assigned to another hepatic condition. To discuss adult presentations of PFIC, the European Medical Journal (EMJ) sat down with Thomas Berg, Head of the Division of Hepatology at Leipzig University Medical Center, Germany. PFIC diagnosis in his centre includes a typical panel of tests for liver diseases along with genetic testing. Symptoms of PFIC can include jaundice, fatigue, and pruritus, with health-related quality of life (HRQoL) severely impacted in some patients, especially if pruritus is present. Treatment for PFIC may be with an ileal bile acid transporter (IBAT) inhibitor or ursodeoxycholic acid (UDCA) to lower serum bile acid levels. There are a number of gaps in the understanding of adult presentations of PFIC. These include the need for epidemiological data, knowledge of the level of misdiagnosed or undiagnosed PFIC in adults, goals of treatment, and which treatment is most suitable. As symptoms of PFIC may be similar to those in other forms of cholestasis, such as primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), or autoimmune hepatitis, to avoid misdiagnosis, analysis of liver enzymes, bile acid levels, and genetic variants of hepato-canalicular transporters should be used to separate PFIC from such conditions.
OVERVIEW
The rare liver disorder PFIC is most frequently subtyped by the affected gene into PFIC1 (ATP8B1, coding for the protein familial intrahepatic cholestasis 1),1 PFIC2 (ABCB11, coding for the protein bile salt exporter pump),2 and PFIC3 (ABCB4, coding for the protein multidrug resistance class III; Table 1).3 More recently, other associated genes have been found, suggesting that there are at least six subtypes of PFIC; however, these are very rare.4 Hepatocellular transport defects and cholestasis in PFIC typically present as jaundice, pruritus, and fatigue. Untreated, PFIC can lead to end-stage liver disease.5,6
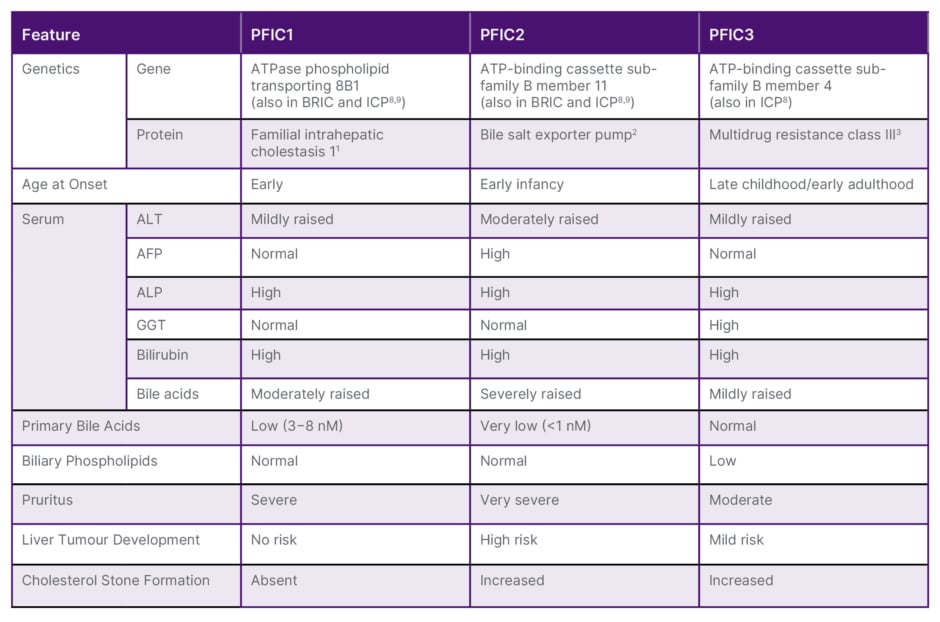
Table 1: Clinical, biochemical, and histological features of different types of progressive familial intrahepatic cholestasis.5-7
AFP: alpha fetoprotein; ALP: alkaline phosphatase; ALT: alanine aminotransferase; ATP: adenosine triphosphate; BRIC: benign recurrent intrahepatic cholestasis; GGT: gamma glutamyl transpeptidase; ICP: intrahepatic cholestasis of pregnancy; PFIC: progressive familial intrahepatic cholestasis.
As well as PFIC subtypes being distinguished genetically, as can be seen in Table 1, there are biochemical differences, such as in levels of alanine aminotransferase, alpha fetoprotein, gamma glutamyl transpeptidase (GGT), and bile acids, and differences in factors such as pruritus severity and the propensity to develop liver tumours.5-7
Although it is being increasingly recognised that PFIC, especially PFIC3, can also first manifest in young adulthood, or even into later life,8 there is little data or discussion around such occurrences. Herein, Berg, an expert in PFIC, discusses some of the issues around recognising, diagnosing, and treating PFIC when it arises for the first time in an adult.
DIAGNOSTIC PATHWAY FOR ADULT PRESENTATIONS OF PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
Suspecting Progressive Familial Intrahepatic Cholestasis
Diagnosis of PFIC is typically carried out at a specialist centre following referral from primary care. Berg described three main reasons a patient might be seen in such a setting, including the one he heads in Leipzig. First, are patients with chronic, progressive liver disease and a suspicion of PFIC, who, he said, “mostly have a long history of elevated transaminase or cholestatic enzymes” and may be “diagnosed at a late stage when they already have cirrhosis.” The second scenario Berg discussed is a young adult with gallstones. “Quite often people do not think about the reason they have gallstones at a very young age, but it could be a sign of a genetic disorder,” he said.
The third scenario is a patient presenting with jaundice and pruritus. The latter symptom can be particularly severe and can heavily impair HRQoL.7 “It is something that affects all parts of your life,” explained Berg, discussing how pruritus can be so bad, a patient may even express suicidality. Of note though, he said that not all of his patients with PFIC experience pruritus.
Figure 1 shows the main hallmarks of PFIC that healthcare professionals should be aware of. Testing for PFIC should include a routine workup for liver diseases with similar symptoms to PFIC, such as PBC, PSC, or autoimmune hepatitis. A suspicion of a genetic cholestatic disease, such as PFIC, should be made if the diagnosis is unclear.4
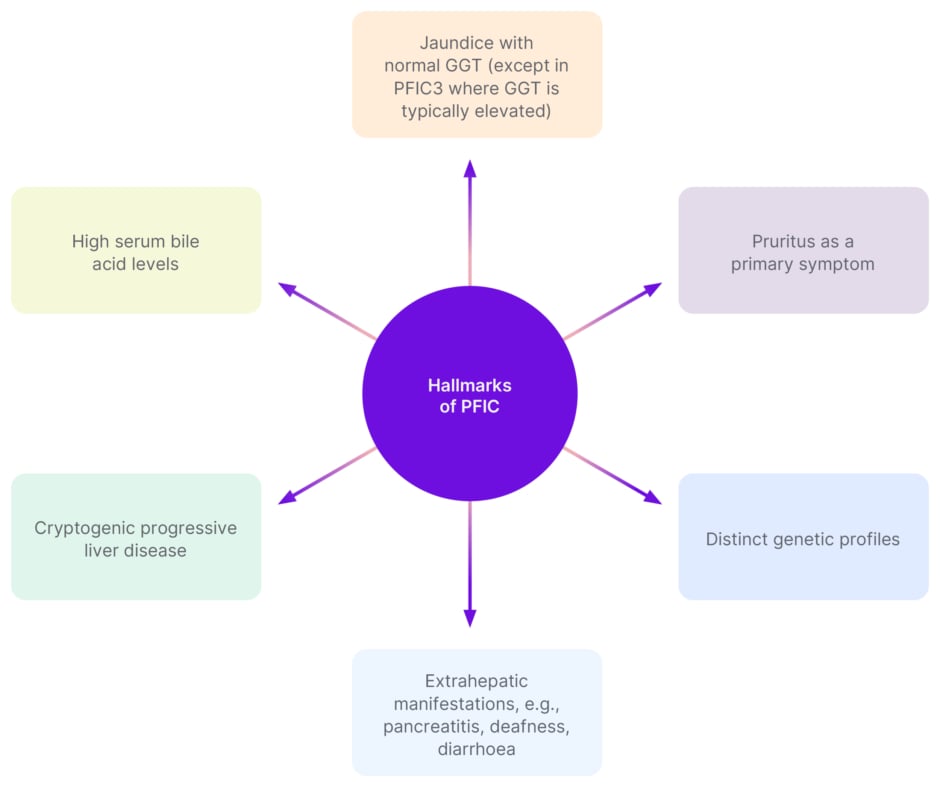
Figure 1: Hallmarks of progressive familial intrahepatic cholestasis.5-7
GGT: gamma-glutamyl transferase; PFIC: progressive familial intrahepatic cholestasis.
Berg described how, in some cases, patients have mildly elevated GGT over the years but “many physicians do not care about two or three times elevated GGT levels.” However, he cautioned: “If this starts at a very young age and really persists, it’s important to do genetic testing for PFIC3.” As an example, Berg highlighted a patient he saw for the first time, aged 65 years, with a “smouldering” form of PFIC. The patient had liver cirrhosis and elevated GGT, which, on examination of their medical history, had first been detected as mildly raised at 18 years old. Genetic testing revealed a PFIC-related pathogenic variant in both the patient and family members, many of whom had mild cholestatic disease, but no elevated bile acids, jaundice, or pruritus.
One reason PFIC may not manifest until adulthood is that, for some, symptoms only arise following a trigger not present in childhood. For example, a PFIC-related hepatic canalicular membrane transporter may function reasonably well under normal conditions but could be overwhelmed in adulthood due to an infection, hormonal changes in pregnancy, or use of a hepatically metabolised medication, at which point clinical symptoms become apparent.8,10
‘Benign’ Recurrent Intrahepatic Cholestasis and Intrahepatic Cholestasis of Pregnancy
There is currently debate around whether two other cholestatic conditions, BRIC and ICP, should be categorised more closely with PFIC, but as episodic forms. BRIC arises due to ATP8B1 and ABCB11 mutations.8,9 Of note, Berg discussed how ‘benign’ may be misleading, and it should simply be referred to as ‘recurrent intrahepatic cholestasis’. “These patients may episodically have very severe jaundice and pruritus that can last for months,”8 Berg explained, “and they are at a higher risk for hepatocellular carcinoma, so need regular surveillance.”9 Another form of cholestasis is one that can arise during pregnancy. This can be due to ATP8B1, ABCB11, or ABCB4 mutations.8 BRIC and ICP are noteworthy as, while changes in bile acid levels during an episode may indicate a PFIC diagnosis (Table 1), levels can be relatively low between episodes.5 As such, it is important to note the dynamic of bile acids when symptoms arise or disappear.
Progressive Familial Intrahepatic Cholestasis Misdiagnosis
As PFIC is a rare disease, physicians who are not in a specialist centre may see it only once in their lifetime and may not know to refer the patient if the symptoms are not severe. Misdiagnosis may occur as PFIC symptoms can be similar to other hepatic conditions. PBC, for example, typically presents in middle-aged females and has anti-mitochondrial antibodies present.11 Therefore in a patient where the histological diagnosis resembles PBC but there are no anti-mitochondrial antibodies, and potentially, they are younger and are male, then a genetic disease might be a more likely suspect. “The same holds true for PSC,” Berg said, which “normally presents at a young age and is associated with inflammatory bowel disease, mostly ulcerative colitis.”12 “If it presents at a later stage, and we have bile duct changes but no inflammatory bowel disease and perhaps no PSC-associated antibodies, then you should suspect PFIC,” he explained.
Genetic Testing
In the above cases where misdiagnosis is a potential, or in others with unexplained cholestatic or liver disease, Berg advised to carry out genetic testing and use the presence of serum bile acids to help aid diagnosis.1-3,5,8,9,13 This currently may not be routinely carried out, even though it is recommended in guidelines.4 Although a genetic test is not needed to make a diagnosis, Berg noted that “we would certainly recommend it because it’s another piece of the puzzle.”
If PFIC presents in childhood, then the genetic correlation may be obvious; however, posited Berg, first presentation in adulthood may mean the severity of the genetic defect, and how the relevant transporter function is affected, may be lower, so a diagnosis of PFIC is missed. It may also be that the typical paediatric genetic markers are not present.1-3 “Sometimes, with multi-smouldering variants, it could be difficult to get clear-cut information by genetic testing,” Berg explained. “If it’s a very mild form, you might find a higher number of variants that are also common in the overall population. This could make diagnosis difficult.”
For adults, there is still a need to understand genotype–phenotype correlation. As an example, Berg discussed two siblings with the same PFIC-linked mutation. One presented with cirrhosis in their early 20s and needed a liver transplant, but the other presented with typical BRIC features with severe recurrent jaundice episodes but no progressive liver disease. This indicates how predicting disease from genetic profile is not always simple.
TREATMENT
PFIC can be treated with an IBAT inhibitor, UDCA, rifampicin, or cholestyramine.4,14 With an IBAT inhibitor, reported Berg, “we hope that by lowering bile acids, because they are toxic, we can help patients in terms of mitigating disease progression.” If bile acids are lowered by at least 70% or to below 70 µmol/L (as was the desired outcome during a clinical trial of the IBAT inhibitor odevixibat),15 there is hope, said Berg, “that this has an effect on disease progression.” Of note though, there are few studies of IBAT inhibitors for PFIC in adults, and Berg described how he treats only a few adult patients where liver disease progression is high enough to justify an IBAT inhibitor prescription. It should also be noted that the effectiveness of IBAT inhibitors can vary depending on underlying genetic variants. Specifically, in a subtype of PFIC2 with a truncating mutation in the ABCB11 gene, these drugs may not significantly impact patients as this leads to severe deficits in the bile salt transport pump. As a result, in this subtype, there is minimal or no bile acid secretion into the gut, rendering IBAT inhibitors ineffective due to the absence of their target substrate.16,17 For patients with relatively mild disease, or with bile duct stones, Berg suggested UDCA with or without fibrates.4
FURTHER RESEARCH
Epidemiological data regarding adult presentations of PFIC is sparse, so cross-sectional studies, and those where all patients with unexplained cryptogenic cirrhosis are checked for PFIC-associated genetic variants, are needed. “We should be more liberal with genetic testing,” urged Berg. “Quite often people check for typical variants that are associated with PFIC, but the more we do research, the more variants we see.” He explained: “We need really in-depth, genetic characterisation to better understand genotype–phenotype correlation; this may help predict disease progression. We must also conduct comprehensive genetic testing using next-generation sequencing to thoroughly investigate the full spectrum of genetic variants and their combinations associated with PFIC disorder.” Berg also discussed how “there might be other genetic factors that may modify the ones we have found.” More data are also needed regarding IBAT inhibitor use in adult presentations of PFIC. Finally, Berg suggested that physicians also need guidance on the best tests to undertake to detect PFIC in adults.
CONCLUSION
While most primary care physicians may rarely see an adult with PFIC, awareness that it can arise beyond childhood is paramount. Referral to specialist care should lead to genetic testing to understand the nature of the cholestasis a patient is experiencing and help guide treatment.4,13 Further studies are needed to understand the epidemiology of PFIC in adults, the phenotype–genotype correlation, and to assess the long-term use of existing therapies on liver outcomes.