Abstract
Hepatic fibrosis is a progressive disease with few treatment options outside of transplantation. Relaxin is a member of the insulin/relaxin superfamily of peptide hormones. Originally known for its roles in pregnancy, relaxin promotes reproductive tissue remodelling and regulates vascular changes, including increased arterial compliance and reduced vascular resistance. Outside of pregnancy, relaxin plays a major role in the protection of organs from excess extracellular matrix accumulation, as demonstrated by the relaxin-null mouse, which develops widespread fibrosis with ageing. Relaxin reduces scarring due to excess collagen deposition by inhibiting collagen production while simultaneously promoting its degradation and can reduce established fibrosis in several animal models of extracellular matrix-associated disease, including liver fibrosis. Treatment with relaxin reduces the myofibroblastic phenotype of activated hepatic stellate cells, the major hepatic collagen-producing cell in fibrosis and cirrhosis. Relaxin also has haemodynamic effects, including vasodilation, and can reduce portal hypertension associated with cirrhosis. In this review, a brief overview of hepatic fibrosis and the role of the hepatic stellate cell will be presented, followed by an introduction to relaxin and its actions. The use of relaxin to treat preclinical models of fibrotic diseases, including liver diseases, will also be discussed. Finally, the completed, current, and ongoing clinical trials of relaxin in human disease will be described, followed by the limitations and future directions for the use of relaxin for disease treatment.
INTRODUCTION
Hepatic fibrosis is a progressive liver condition resulting from many causes of chronic liver injury, including viral and autoimmune hepatitis, nonalcoholic fatty liver disease (NAFLD), steatohepatitis, and alcoholic liver disease.1 The production of fibrillary collagen is necessary for the normal response to injury. Following acute damage, injury-related collagen is rapidly removed, and the liver structure and function are restored; however, with chronic disease, unabated collagen production leads to fibrosis, which disrupts liver architecture and function. Chronic liver disease affected 29 million individuals in Europe and 30 million in the USA in 2013.2,3 Chronic liver disease often progresses to cirrhosis, a condition that caused 37,000 deaths in the USA, making it the 12th leading cause of death in the USA in 2013, and caused 170,000 deaths in Europe in 2013.2,3 Currently, treatment options for hepatic fibrosis and cirrhosis are limited to the removal of the injurious stimuli, implementation of antiviral approaches, or organ transplantation.1 Therefore, new approaches toward treatment of these conditions are sorely needed.
HEPATIC STELLATE CELLS
Chronic liver injury leads to hepatic fibrosis, characterised by excess extracellular matrix deposition, loss of liver function, and ultimately cirrhosis and liver failure.4 The increased matrix formation is not only due to accelerated synthesis and secretion of collagen and other extracellular matrix components, but also to decreased clearance of matrix by matrix metalloproteinases (MMP).1 This is a co-ordinated response mediated by interactions between the hepatocytes and the nonparenchymal cell population, including the sinusoidal endothelial cells, resident macrophages (Kupffer cells), and the hepatic stellate cells (HSC).
Under normal conditions, the HSC are perisinusoidal cells that resemble both fibroblasts and lipid-storing cells. In the absence of injury, HSC display a quiescent phenotype and function primarily to store lipids in the form of retinoids.5 A major event in the response to liver injury is the transactivation of HSC resulting in an activated myofibroblastic phenotype. In this state, the stored retinoids are released. HSC begin producing large amounts of extracellular matrix, including collagen Types I and III, and secrete tissue inhibitors of metalloproteinases (TIMP), which serve to decrease matrix degradation.6 Therefore, HSC are the major source of extracellular matrix in liver fibrosis. Activated HSC are also characterised by increased proliferation and sensitivity to cytokines, expression of smooth muscle actin, and increased contractility in response to endothelin-1.6 Upon removal of the cause of injury, the activated HSC are removed, either by apoptosis or regression to the quiescent phenotype. However, in chronic disease, the activated HSC continue to proliferate and produce excess extracellular matrix; activated HSC are therefore a prime target for antifibrotic therapy. Ideally, an agent that targets activated HSC would inhibit the activated phenotype and promote matrix clearance to remove the scarring; one such candidate agent is the hormone relaxin.
RELAXIN: AN ANTIFIBROTIC HORMONE
Relaxin is a polypeptide hormone of the insulin/relaxin superfamily.7 The first identified function for relaxin was its role in pregnancy, during which it is produced by the corpus luteum of the ovary. During pregnancy, circulating relaxin levels in humans peak during the first trimester, but there are variable release patterns in different species.8 Relaxin is involved in a number of critical events associated with pregnancy, including softening of the cervix and vagina.8 Relaxin also has potent vascular effects and plays a major role in increasing arterial compliance and reducing vascular resistance, while at the same time increasing renal plasma flow and glomerular filtration rate.9,10 There is also evidence that relaxin may have vascular effects in males and nonpregnant females, but these results are inconsistent and require further study.10
An important mechanism associated with many of the effects of relaxin is the widespread remodelling of extracellular matrix, involving both the altered secretion of extracellular matrix components as well as changes in matrix component degradation.11 These observations have been extended to nonreproductive tissues, where a role has emerged for relaxin as a general antifibrotic hormone.12 Relaxin has been shown to reduce collagen production, increase MMP activity, decrease the levels of TIMP in scleroderma cell models, and decrease experimentally induced dermal fibrosis in rodents.13,14 Similarly, relaxin was effective in preventing or reversing several models of experimental renal fibrosis in rodents, and the hormone-modulated collagen, MMP, and TIMP levels in renal fibroblasts.15 Relaxin decreased the myofibroblastic phenotype of lung fibroblasts and decreased pulmonary fibrosis in rodents.11 Finally, numerous studies have implicated relaxin in the prevention or treatment of several models of cardiac fibrosis.11
The hypothesis that relaxin has a role as a general protective agent against fibrosis was strengthened by observations made using the relaxin-null mouse. In addition to the expected difficulties with reproduction, these mice spontaneously developed age-related pulmonary, cardiac, dermal, and renal fibrosis, which was reversible with relaxin treatment.16 In most cases, male mice developed more severe fibrosis than the females, likely due to testosterone-related effects.17 Similar effects were seen in mice lacking Rxfp1, which codes for the cognate receptor for relaxin. These Rxfp1 mice developed sex-specific pulmonary fibrosis with age.18,19
ROLES FOR RELAXIN IN THE LIVER
Relaxin also has effects in the liver. Rats that received relaxin treatment demonstrated acute changes in hepatic microcirculation; morphological changes were detected in sinusoidal myofibroblastic cells lacking retinoids, most likely activated HSC.20,21 In addition, the relaxin-null mouse presented with increased liver weight.22 Relaxin treatment of activated rat HSC decreased overall collagen and TIMP production.23 Similarly, treatment of activated mouse HSC resulted in decreased total collagen deposition, collagen synthesis, Type I collagen secretion, and reduced smooth muscle actin expression, but had no effect on HSC proliferation or apoptosis.24 Relaxin also promoted a matrix-degrading phenotype inhibiting TIMP secretion and, by increasing the expression and activity of MMP13, the major rodent fibrillary collagen-degrading MMP.24 The expression of the relaxin receptor, RXFP1, was found to be low or undetectable in quiescent mouse HSC or normal liver but increased dramatically upon HSC activation or development of hepatic fibrosis.25 This finding was later confirmed using rat and human activated HSC and tissues,26 providing evidence that the activated HSC are a target of the antifibrotic actions of relaxin.
Consistent with the findings using cultured cell models, relaxin has shown effectiveness in in vivo models of experimental hepatic fibrosis (summarised in Table 1). While the effects of relaxin were favourable overall, there were differences dependent on the dose and timing of treatment. In a rat prevention model, in which treatment with recombinant human relaxin (infusion of 0.5–0.6 mg/kg/day) began concomitantly with carbon tetrachloride (CCl4)-induced liver injury for 4 weeks, relaxin decreased total hepatic collagen content.23 In a similar prevention study using mice, infusion of porcine relaxin (0.5 mg/kg/day) reduced serum liver transaminases, hepatic collagen, and smooth muscle actin content.27 In a second study using a more clinically relevant mouse model, fibrosis was first established with 4 weeks of CCl4 followed by porcine relaxin infusion (0.5 mg/kg/day) for 1 or 2 weeks with continued CCl4 administration.27 After 1 week of relaxin treatment, hepatic collagen and smooth muscle actin content were modestly decreased, but after 2 weeks only the smooth muscle actin content was significantly reduced.27 In a later study, mice were made fibrotic by CCl4 treatment for a total of 8 weeks, with relaxin treatment for the final 4 weeks at lower relaxin doses (25 or 75 µg/kg/day infusion of recombinant human relaxin, also known as serelaxin).28 The lower doses were used due to evidence that extended treatment of human relaxin at high doses can induce an immune response in rodents.34 In this model, relaxin at either dose reduced hepatic smooth muscle and collagen content, and at the higher dose the expression of Type I procollagen, TIMP2, and MMP2 was decreased, while expression of MMP3 and MMP13, and overall Type I collagen-degrading activity was elevated.28 Another study examined the effect of short-term treatment (72 hours infusion of 0.5 mg/kg/day recombinant human relaxin) on rats with either established fibrosis (8 weeks CCl4 treatment), cirrhosis (16 weeks CCl4 treatment), or cholestatic disease induced by surgical ligation of the common bile duct.26 Short-term relaxin infusion had no effect on total collagen content in any of the models but decreased smooth muscle actin in the 8 week fibrosis model. However, in all three models, relaxin significantly decreased portal blood pressure without decreasing systemic blood pressure, providing evidence that relaxin may be effective in reducing the intrahepatic vascular resistance characteristic of cirrhosis. Finally, adenoviral delivery of relaxin was trialled in a thioacetamide model of hepatic fibrosis.29 Rats were treated for 8 weeks with thioacetamide, then infected with adenovirus after Week 7 and examined either 3 days or 3 weeks later.29 No significant effects were observed 3 days after infection, but after 3 weeks the adenovirally-delivered relaxin reduced liver collagen and smooth muscle actin content and decreased gene expression of Type I collagen and TIMP2. In summary, these preclinical studies suggest that prolonged relaxin treatment may be effective in treating hepatic fibrosis, while short-term treatment may be useful to reduce portal blood pressure.
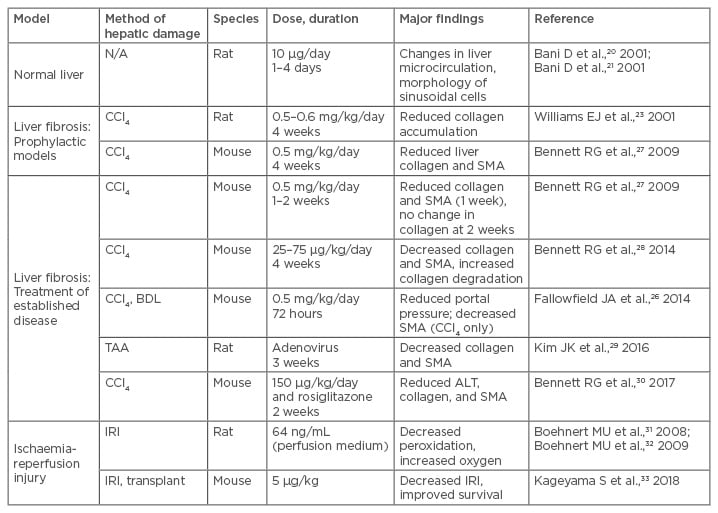
Table 1: Summary of preclinical studies investigating relaxin based therapeutics in liver disease.
ALT: alanine transaminase; BDL: bile duct ligation; CCl4: carbon tetrachloride; IRI: ischaemia-reperfusion injury; N/A: not applicable; SMA: smooth muscle actin; TAA: thioacetamide.
CROSSTALK BETWEEN RELAXIN AND PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR GAMMA
A second critical regulatory element in HSC activation is the peroxisome proliferator-activated receptor gamma (PPARγ) pathway. PPARγ is a transcription factor activated by antidiabetic thiazolidinedione (TZD) drugs and some prostaglandins.35 Expression of PPARγ is detectable in quiescent HSC but is lacking in activated HSC and myofibroblasts.6 Restoration of PPARγ expression, either by treatment of activated HSC with PPARγ ligands or by forced expression of PPARγ, induced a reversion of the HSC to a state that closely resembled the quiescent phenotype, as shown by decreased proliferation; reduced SMA, collagen, and TIMP expression; increased MMP-13 expression; and restoration of lipid storage.36 Treatment of experimentally induced fibrosis with PPARγ ligands prevented hepatic fibrosis in vivo.37 These findings led to great interest in the use of TZD for the treatment of hepatic fibrosis because the quiescent HSC have adipogenic properties and PPARγ is a major inducer of adipogenesis. However, recent studies have suggested that TZD treatment may be ineffective for established fibrosis in rodents, casting some doubt on the use of using TZD alone for this purpose.38,39
There is recent evidence that relaxin signalling through RXFP1 results in activation of PPARγ. In cultured HSC, relaxin caused increased transcriptional activity of PPARγ.40 Interestingly, the mechanism was ligand-independent, as inhibitors of PPARγ ligand binding did not block the effect.40 Relaxin was also shown to activate PPARγ in cerebral arterioles.41,42 More recently, the mechanism for this stimulation was found to be via increased expression of a coactivator protein, known as PPARγ coactivator 1 alpha, via cAMP and p38-MAPK-dependent pathways.43 Therefore, relaxin may enhance PPARγ activity by inducing the cAMP/protein kinase A pathway. Other activators of protein kinase A enhanced basal and ligand-induced PPARγ transcriptional activity and enhanced the stability of PPARγ-DNA binding in fibroblasts and adipocytes.44,45 This raises the possibility that relaxin may sensitise cells to respond to TZD drugs. This was addressed in a recent study that used a combination of relaxin and the TZD drug rosiglitazone on established hepatic fibrosis in mice.30 The results confirmed that rosiglitazone alone is ineffective but found that the combination of relaxin and rosiglitazone was more effective than either treatment alone.30 This notion is consistent with the recent finding that the response to airway dilators, including rosiglitazone, was increased when used in combination with relaxin.46
CURRENT AND POTENTIAL CLINICAL STUDIES OF RELAXIN
The majority of the human studies not focussing on reproduction have focussed primarily on the haemodynamic properties of relaxin. Most of these have examined the treatment of heart disease, based on the effects of relaxin on vasodilation and improved renal function.47 A large Phase III randomised, placebo-controlled, double blind trial (RELAX-AHF) was conducted involving 1,161 patients presenting with acute heart failure.48 Study participants were given a 48 hour infusion of recombinant human relaxin (30 µg/kg/day) or placebo, with the primary endpoints of improved dyspnoea by Likert scale or visual analogue scale. One primary end point was reached (improvement in dyspnoea identified through the visual analogue scale) but not the other. The study found a significant decrease in all-cause mortality and cardiovascular death at 180 days in the relaxin-treated subjects. Failure to secure approval from the U.S. Food and Drug Administration (FDA) led to a second, larger Phase III trial (6,566 participants), known as RELAX-AHF-2, with primary endpoints of reduced cardiovascular mortality and worsening heart failure at 180 days.49 Although the results have not been published at the time of writing, the sponsor has issued a press release stating that the primary endpoints were not met.50
The clinical studies of relaxin for human fibrotic disease have thus far been limited to scleroderma. The earliest studies were conducted in the 1950s, using porcine relaxin to treat scleroderma.11 The results from these studies were highly variable, and were not followed up until more recent trials were conducted using recombinant human relaxin. After promising results in a Phase II study, a larger Phase III study revealed no significant effects after continuous infusion with 10 or 25 μg/kg/day relaxin for 24 weeks, possibly due to the advanced stage of the disease in many of the subjects.51 Indeed, relaxin treatment failed to reverse advanced dermal fibrosis in the aged relaxin-null mouse, suggesting that severe scleroderma may be irreversible.52 It is possible that further studies of relaxin treatment in subjects with less severe scleroderma might yield more positive results. Another possible factor may be reduced expression of RXFP1 in skin from scleroderma patients.53
To date, the clinical studies of relaxin for liver disease have focussed on reducing portal hypertension. As described previously, preclinical models suggest that a short infusion of relaxin resulted in reduced portal blood pressure without reducing systemic blood pressure. In a Phase II, open label study of 40 patients with cirrhosis and hypertension, two consecutive 1 hour infusions (1 hour of 80 µg/kg/day followed by 1 hour of 30 µg/kg/day) recombinant human relaxin resulted in increased renal arterial flow, with no significant effect on systemic blood pressure or hepatic perfusion, and the treatment was well-tolerated.54 A second randomised placebo-controlled study Phase II trial is being prepared.55 The study will analyse 2 hours of relaxin infusion in patients with cirrhosis and portal hypertension, with the primary endpoint of change in fasting hepatic venous pressure gradient.
Finally, there is evidence that relaxin may act as a hepatic protective agent from ischaemia-reperfusion injury and therefore act as a preservative for liver transplantation tissue. Relaxin added to organ preservation solution for rat liver perfusion decreased markers of oxidative damage and increased the hepatic oxygen supply.31,32 Recently, a study examining a mouse model of orthotopic liver transplantation after 18 hours cold storage found that relaxin decreased ischaemia-reperfusion damage and improved survival.33 Therefore, it is possible that relaxin may act to improve the viability of human livers for transplantation.
PERSPECTIVE AND LIMITATIONS
The preclinical studies suggest that relaxin is an effective treatment for a wide variety of fibrotic disease models, including hepatic fibrosis and cirrhosis-related portal hypertension. Thus far, no human studies of relaxin treatment of fibrotic disease have been attempted, apart from scleroderma. This may partly be the result of the failure of the Phase III scleroderma study casting doubt for future long-term studies using relaxin. However, given the severity of the disease and the questions about the relevance of the clinical outcomes used,56 further studies are needed. Additionally, with the recent failure of the RELAX-AHF-2 study, the future of major clinical studies using recombinant human relaxin is unclear.
One major limitation for the extended use of recombinant human relaxin (or relaxin derived from any species) is its nature as a peptide hormone. This property makes relaxin unstable, with a relatively short half-life (approximately 10 minutes) in circulation.57 Furthermore, as a peptide, relaxin is unsuitable as an oral agent, and so the hormone must be administered intravenously by frequent injections or by continuous subcutaneous delivery. For these reasons, there is much attention currently aimed at producing modified relaxin-based peptides with improved stability.57,58 Additionally, as described above, preclinical and clinical studies have used a wide range of relaxin doses and treatment durations, with varying degrees of success. A contributing factor may be related to the ‘bell-shaped’ responses to relaxin that have been frequently observed.59 Additional studies are needed to systematically optimise relaxin doses and treatment durations for each target disease.
Small molecule activators of RXFP1 may play an important role in the future use of the relaxin signalling pathway in the treatment of human disease. The best characterised of these small molecule RXFP1 agonists is ML290, which is much more stable than relaxin, and is currently serving as the lead compound in optimisation studies.60,61 In animal models, ML290 has shown properties similar to relaxin in reproductive tissues and cardiac function and has had relaxin-like effects on HSC.62 ML290 is a promising lead compound for the development of future small molecule relaxin-mimetic drugs.
Another limitation that may impact the successful clinical application of relaxin relates to tissue expression of RXFP1 in disease states. In the liver, the expression of RXFP1 increases in activated HSC in preclinical models and human disease, supporting the use of relaxin in hepatic fibrosis.25,26 However, in human scleroderma and idiopathic pulmonary fibrosis, expression of RXFP1 appears to be reduced, suggesting that the ability to respond to relaxin may be reduced in these diseases.53,60 Additional studies of disease-specific RXFP1 expression are needed to inform the applicability of relaxin to these states.
The interaction of relaxin-RXFP1 signalling with other pathways, such as PPARγ, suggest that combination treatment of relaxin and agents such as the TZD drugs may present an improvement over monotherapy.30,43 Indeed, crosstalk between relaxin and other pathways, such as the angiotensin II type 2 receptor (AT2R), has been demonstrated. It was shown that RXFP1 can heterodimerise with AT2R and that AT2R activity was necessary for the antifibrotic effect of relaxin in a model of renal fibrosis, suggesting that simultaneous targeting of RXFP1 and AT2R may be a promising avenue of investigation.64
CONCLUSION
In summary, relaxin has shown great promise as an antifibrotic agent in cell culture and animal models of fibrosis. The role of relaxin in the protection from excess collagen accumulation is clear from the studies of relaxin-null and RXFP1-null mice, which developed spontaneous fibrotic diseases with ageing. Due to its antifibrotic and haemodynamic properties, as well as the limited expression profile of RXFP1, the relaxin signalling pathway remains a promising target for the development of new treatment options for liver diseases.