Abstract
Raised levels of the blood coagulation protein von Willebrand factor (VWF) are now recognised to be important in patients with liver disease. The markedly raised plasma VWF levels in patients with acute liver failure and acute-on-chronic liver failure may contribute to the pathogenesis of liver failure, and of multi-organ failure, by impeding microcirculatory perfusion in the liver and the other affected vital organs. In this review, the authors present a brief introduction to VWF biology, discuss the ability of raised plasma VWF levels to accurately predict survival in different syndromes of liver diseases, speculate why plasma VWF levels are raised in liver failure syndromes, and examine the therapeutic potential of VWF-lowering therapies in these scenarios.
INTRODUCTION
The most commonly inherited bleeding disorder, von Willebrand disease, is caused by the deficiency of von Willebrand factor (VWF). In contrast to von Willebrand disease, the opposite scenario of raised VWF levels in circulation is attracting increasing attention in patients with liver diseases. The VWF protein has binding sites for platelets as well as factor VIII and collagen, thus it has important roles in both primary and secondary haemostasis.
While VWF deficiency is associated with bleeding tendency, raised VWF levels are a minor risk factor for thrombotic events such as cerebrovascular accidents and coronary artery disease.1,2 Recent reports have documented mild increases of plasma VWF levels in patients with cirrhosis, as well as a marked increase of plasma VWF levels in patients with acute and acute-on-chronic liver failure (ACLF). The raised plasma VWF levels are relatively accurate in predicting survival in patients with liver disease (cirrhosis, ACLF, and in acute liver injury and failure).
AN INTRODUCTION TO VON WILLEBRAND FACTOR BIOLOGY FOR THE HEPATOLOGIST/GASTROENTEROLOGIST
von Willebrand Factor Synthesis, Storage, and Secretion
VWF is a large, adhesive glycoprotein synthesised by endothelial cells and megakaryocytes. The VWF protein is stored in Weibel–Palade bodies in endothelial cells and in α-granules of platelets. Secretagogues, such as vascular endothelial growth factor (VEGF), histamine, thrombin, oestrogen, and desmopressin, and inhibitors, such as nitric oxide and dopamine, influence VWF secretion via endothelial cells and platelets.3
The different forms, or multimers, of VWF exist as low, intermediate, high, and ultra-large-molecular-weight forms. In endothelial cells and platelets, VWF is stored as ultra-large multimers. As the secreted VWF rapidly undergoes proteolysis, the size of ultra-large VWF multimers is reduced; therefore, typically the ultra-large multimers of VWF are not found in plasma. Constitutively secreted forms of VWF are shorter and are usually high-molecular-weight forms. Circulating plasma has high (large), intermediate, and low sized VWF multimers. The size of VWF multimers determines its haemostatic function. Of the different sized VWF multimers in circulation, the high-molecular-weight fraction has the most potent haemostatic function.4
Some of the endothelial cell-derived VWF multimers remain bound onto the endothelial cell surface. The globular form of VWF is stretched out under the high shear stress of normal blood flow (such as in arterioles) to a string-like form. In these stretched-out, string-like forms of VWF, the site for cleavage of VWF by ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin Type 1 motif, member 13) is exposed.
Three types of VWF are recognised in the human body: soluble plasma VWF, basement membrane VWF (i.e., extracellular matrix VWF), and cellular VWF. VWF is the largest known protein in human plasma. The high-molecular-weight-forms of VWF have molecular weights of up to 10,000 kDa.4
von Willebrand Factor Function
VWF is an important blood coagulation protein. The three main haemostatic functions of VWF are to mediate platelet–platelet interactions, platelet–subendothelial collagen interactions in the vessel wall, and to be a carrier of factor VIII in circulating blood. Recently, nonhaemostatic functions of VWF have also been proposed, such as roles in angiogenesis, smooth muscle cell proliferation, tumour cell metastasis, and immune cell regulation.3
VWF protein has separate sites, or domains, for binding to platelets and collagen. Collagen located in the subendothelial space is exposed when a vessel wall is injured. VWF binds to collagen, thus exposed. VWF, bound to the collagen, localises factor VIII (VWF functions as a factor VIII carrier). The platelet-binding domain of the VWF protein is exposed when VWF is unfolded and stretched out by the shear stress of arterial circulation. Thus, platelets are recruited to the site of vascular injury.
As described above, the VWF molecule has a vital role both in primary haemostasis (platelet plug formation), as well as in secondary haemostasis (in the coagulation cascade). The binding activity of VWF to collagen is used to assay VWF activity; this is called VWF collagen binding activity. The binding activity of VWF to platelets can also be used to assay VWF activity, as measured by the ristocetin cofactor activity assay.
HOW TO ASSAY VON WILLEBRAND FACTOR LEVELS IN PLASMA
The assays for VWF have been mostly developed and tested with the aim of diagnosing VWF deficiency (i.e., to diagnose and to accurately subtype von Willebrand disease).5
Plasma samples for assays of VWF require several specific precautions to be undertaken during sample collection and processing. A clean venepuncture is advised, with minimal venous stasis, and a 19–21-gauge needle. The VWF assays are performed on platelet-poor plasma; for example, the blood sample is collected using 0.109 M citrate anticoagulant and centrifuged at 2,500 g for 15 mins, and separated plasma is used for VWF assays. In patients with liver disease, it is important to obtain a baseline blood sample for VWF assays prior to administration of any VWF-lowering treatments, such as N-acetylcysteine, fresh frozen plasma, or therapeutic plasma exchange.
VWF antigen is measured using an ELISA kit. Activity of VWF is assayed as collagen binding activity6 and as ristocetin cofactor activity,7 which reflects its binding affinity for collagen and platelets, respectively. The multimeric structure of VWF may be determined by agarose gel electrophoresis, followed by Western blotting.8
It has been suggested by Favaloro9 that VWF activity in a collagen binding assay reflects the presence of high-molecular-weight VWF multimers.
RAISED PLASMA VON WILLEBRAND FACTOR LEVELS AND MULTI-ORGAN FAILURE IN CRITICALLY ILL PATIENTS
Thrombo-Inflammation
The close relationship between inflammation and thrombosis, especially in primary haemostasis, has recently been the subject of active research. ‘Thrombo-inflammation’, secondary to the co-ordinated activation of the immune system (such as complement components, or innate immune cells) and primary haemostasis (such as platelets and endothelial cells), is the body’s primary response to a variety of external, potentially harmful stimuli.10 This response is needed for tissue repair after initial damage. The complement components, specifically the lectin and alternative pathways, form the major interface between inflammation and primary haemostasis.11 If not controlled, these responses may lead to widespread thrombotic microangiopathies and inflammatory complications. Thrombotic microangiopathy accompanies sepsis syndrome and has been termed thrombocytopenia-associated multi-organ failure.12
Endothelial Activation in Inflammatory Conditions/Sepsis
In inflammatory conditions, endothelial activation leads to a progressive rise in plasma VWF, especially the ultra-large VWF multimer subset. This is accompanied by a parallel steady decrease in the cleaving protease ADAMTS13, leading to VWF–ADAMTS13 imbalance, which has been shown to contribute to and correlate with the severity of inflammation and organ failure in sepsis.13,14
In a randomised controlled trial involving children with sepsis, reversing the VWF–ADAMTS13 imbalance by therapeutic plasma exchange reversed organ dysfunction in patients with thrombocytopenia-associated multi-organ failure. All seven children who died of thrombocytopenia-associated multi-organ failure had reduced plasma ADAMTS13 levels and VWF-rich microvascular thrombi at autopsy.15
The imbalance of high VWF and low ADAMTS13, and its correlation with disease severity, has been studied in a variety of critical illnesses, including sepsis,16 acute pancreatitis,17 severe malaria,18 dengue fever,19,20 sickle cell disease,21 alcoholic hepatitis,22 and complications such as acute lung injury23 and disseminated intravascular coagulation.24
Plasma von Willebrand Factor Levels as a Prognostic Marker in Critical Illnesses
Multiple studies have explored the role of plasma VWF levels in predicting short-term prognosis in critical illnesses. For example, Hyseni et al.25 found measurement of active VWF plasma levels useful as an independent predictor of short-term outcome in patients with sepsis and systemic inflammatory response syndrome.25
Dynamic changes in plasma VWF levels predicted multiple organ dysfunction and death in patients with scrub typhus,26 and acute lung injury and death in patients with acute pancreatitis.27
The imbalance of high VWF and low ADAMTS13 in sepsis/inflammatory conditions can be a predisposition to platelet microthrombi and an impedance to vital organ microcirculation, leading to multi-organ failure and death in critically ill patients. As multi-organ failure is seen in critically ill patients with a severe grade of ACLF or with acute liver failure, it is possible that raised plasma VWF levels may contribute to disease pathogenesis in these patients.
RAISED PLASMA VON WILLEBRAND FACTOR LEVELS PREDICT SURVIVAL IN PATIENTS WITH CIRRHOSIS, ACUTE-ON-CHRONIC LIVER FAILURE, AND ACUTE HEPATOTOXICITY
VWF was previously recognised as an endothelial activation marker in patients with cirrhosis.28,29 Recent studies document raised plasma VWF levels as a prognostic marker of outcome in patients with acute and chronic liver disease. It is more relevant in liver disease because ADAMTS13 is produced in liver stellate cells. The level of plasma ADAMTS13 has been shown to decrease with increasing severity of liver disease.30,31 This leads to an exaggerated imbalance of primary haemostasis that favours clotting. This imbalance is probably maximised in the presinusoidal portal vein radicles (because ADAMTS13 is secreted from the stellate cells), which, in turn, explains its occurrence in pure vasculopathy in idiopathic noncirrhotic portal hypertension. This progressive imbalance may also play a part in cirrhosis disease deterioration by contributing to portal micro-occlusions and parenchymal extinction.
In patients with idiopathic noncirrhotic intrahepatic portal hypertension due to chronic portal microangiopathy, deficiency of ADAMTS13 is noted despite preserved hepatocellular function.32-35 In patients with noncirrhotic portal hypertension, low plasma ADAMTS13 levels are also associated with vascular complications such as portal vein thrombosis,36 pulmonary hypertension,37 and hepatopulmonary syndrome.38
Plasma von Willebrand Factor Levels Predict Survival in Cirrhosis
In a recent systematic review of five studies on plasma VWF levels in patients with cirrhosis (N=715 patients), Eidelberg et al.39 found that baseline plasma VWF antigen level was an independent predictor of medium-term mortality in patients (median: 25.6; range: 23.6–33.0 months), with an area under the curve of 0.74 (95% confidence interval: 0.70–0.79) and an optimal cut-off median (318%; range: 216–390%).39 Newer reports continue to corroborate these findings.40
Model for end-stage liver disease (MELD) scores, although a good predictor of hepatocellular dysfunction, may not adequately predict portal hypertensive complications, for which a vascular/endothelial marker may be more efficient. A combination of MELD score and VWF antigen levels appears to be a better predictor of survival in patients with cirrhosis. While plasma VWF antigen level was an independent predictor of 3-month mortality in patients listed for liver transplant comparable to MELD-Na, a composite score (VWF antigen and MELD-Na) improved prediction of 3-month waiting list mortality.41
Plasma von Willebrand Factor Levels Predict Survival in Acute Hepatotoxicity
Acute liver failure is often associated with increased release of endotoxins and cytokines, which may lead to a decrease in ADAMTS13 activity and a concomitant rise in VWF levels.42
A mechanistic link between VWF, platelet accumulation in the liver, and liver repair has been suggested in experimental models of acute liver injury.43 In an animal study, low-dose dimethylnitrosamine activated hepatic stellate cells, and influenced the levels of ADAMTS13.44
High plasma levels of VWF and low ADAMTS13 were shown in 50 patients with acute liver injury and acute liver failure. Low baseline ADAMTS13 levels predicted transplant-free survival in these patients. Plasma VWF antigen levels did not predict survival in these patients. However, this study included patients with nine different aetiologies of liver disease. Additionally, the majority of these patients had blood samples drawn for a VWF assay after they had received N-acetylcysteine, as part of their acute liver failure management (N-acetylcysteine may reduce VWF levels).45
In contrast to the above-mentioned study, plasma VWF levels accurately predicted inpatient survival in patients with acute hepatotoxicity caused by a single aetiology (yellow phosphorus contained in rodenticides). In a recent report of 24 patients with rodenticide-induced hepatotoxicity, of whom 20 had acute liver injury, three had acute liver failure and one had uncomplicated acute hepatitis. Plasma VWF antigen levels were raised to a median of 423% (range: 146–890%) in patients with acute liver injury and to a median of 448% (range: 414–555%) in patients with acute liver failure. Normal plasma VWF antigen levels were 50–150%. The raised plasma VWF levels predicted survival more accurately than the MELD and sequential organ failure assessment (SOFA score) in these patients.46
Plasma von Willebrand Factor Levels Predict Survival in Acute-on-Chronic Liver Failure
Plasma VWF levels are noted to be consistently higher in patients with acute-on-chronic liver failure (ACLF).47,48 Prasanna et al.,49 in a study of 50 ACLF patients, showed that plasma VWF levels were 5–7-fold elevated, which correlated independently with organ failure and in-hospital survival. Similarly, in patients with cirrhosis and superimposed inflammation, VWF was high and predicted transplant-free survival.50
WHY ARE PLASMA VON WILLEBRAND FACTOR LEVELS INCREASED IN PATIENTS WITH LIVER FAILURE?
Plasma VWF levels are increased 2–3-fold in patients with chronic liver diseases (cirrhosis patients in the outpatient setting39 and patients with noncirrhotic portal hypertension),34,35 4.0–4.5-fold in patients with acute hepatic dysfunction (acute liver injury and acute liver failure),46 and 5–7-fold in patients with ACLF.39,49 This raises many questions on the reason this occurs; is it because of increased VWF production/secretion, reduced VWF clearance from the circulation, or both? Increased VWF secretion from endothelial cells may reflect endothelial activation. The highest levels of plasma VWF noted in ACLF may reflect acute-on-chronic endothelial activation.51
The Liver is an Important Site of von Willebrand Factor Clearance
Is VWF clearance affected in patients with liver failure? The mechanisms of VWF clearance seem to be linked to adequate liver function. When radiolabelled VWF was injected intravenously into VWF-deficient mice to study biodistribution of VWF, the bulk of VWF protein was noted to be taken up by the liver. When expressed in relative terms, the liver and spleen were equally efficient in VWF uptake.52
Mechanisms of von Willebrand Factor Clearance
Clearance of VWF is performed by macrophages, endothelial cells, and hepatocytes via C-type lectin domain family-4 (CLEC4M) receptors,52 galactose-type lectin receptors,53 low density lipoprotein receptor related protein-1 (LRP-1),54 and Ashwell–Morell receptor.54
The sugar moieties in the VWF glycoprotein seem to influence VWF clearance.55 The glycosylation profile of VWF affects plasma VWF levels.56 VWF protein contains 10 sites for O-linked and 12 sites for N-linked glycosylation. Some of the N-linked sugars contain ABO blood group determinants, which are absent on the O-linked sugars. ABO blood groups appear to influence plasma VWF levels: mean VWF levels are approximately 25% lower in individuals with blood group O compared to non-O individuals; these levels are even further reduced in individuals with the Bombay blood group (who do not express ABO antigens).57
Desialylation of VWF is yet another effect on VWF glycosylation, which leads to its rapid clearance by hepatocytes. Sialylation is important to prevent premature clearance by receptors which recognise non-sialylated terminal galactose residues, such as the Ashwell–Morell receptor expressed on hepatocytes. The purified normal factor VIII/VWF protein possesses both procoagulant activity and ristocetin-induced platelet-aggregating activity, and contains 154±15 nmol of sialic acid/mg of protein and 28±3 mol of sialic acid/mol of 200,000 molecular weight subunits. Desialylation of this protein was associated with reduction in ristocetin-induced platelet aggregating activity, while procoagulant activity, measured by partial thromboplastin time, remained unchanged. Desialylated factor VIII/VWF protein is rapidly cleared in the liver.58
Cleavage of VWF by ADAMTS1359 and by plasmin60 is another mechanism of VWF clearance. Globular forms of VWF are resistant to cleavage by ADAMTS13 and by plasmin in static conditions, though they are readily cleaved under shear stress.
More than 90% of tissue-resident macrophages in the body are located in the liver, known as Kupffer cells. The Kupffer cells are located in hepatic sinusoids and adhere to the hepatic sinusoidal endothelial cells. Kupffer cell population is severely decreased in the initial 24 hours of injury in mouse models of acute liver failure (induced by paracetamol) and chronic liver injury (induced by carbon tetrachloride).61 The cells that synthesise ADAMTS13 (hepatic stellate cells) are located in the perisinusoidal space in the liver.
Hepatic Sinusoidal Endothelium: A ‘von Willebrand Factor-Free’ Zone
VWF is heterogeneously expressed in endothelial cells throughout the vascular tree, with higher expression in veins compared with arteries and arterioles. However, immunostaining and messenger RNA expression studies show that VWF is not expressed on the hepatic sinusoidal endothelial cells in a healthy liver.62 Of note, ADAMTS is produced by hepatic stellate cells.63 Tissue-resident macrophages (>90% are located in hepatic sinusoidal lining) are involved in VWF clearance. Thus, in the healthy liver, hepatic sinusoidal endothelium appears to be a ‘VWF-free’ zone, and an important site for removing VWF from the circulation. It is perhaps unsurprising that VWF levels are raised in different forms of liver disease.
ROLE OF VON WILLEBRAND FACTOR IN PROGRESSION OF LIVER DISEASE
The role of VWF in causing progression of acute liver injury was studied in a mouse model using genetic and antibody-mediated strategies to achieve VWF reduction. Paracetamol overdose-induced acute liver injury in this model was associated with VWF deposition in the liver, raised plasma VWF levels, and impaired VWF clearance. Administration of VWF to VWF-deficient mice led to a delay in the repair of paracetamol-induced liver injury (associated with platelet deposits and increased necrosis in the liver). Interventions to reduce VWF led to reduction in platelet deposition, less necrosis, and accelerated repair of the liver, even in mice with established liver injury. Thus, this experimental model provides clear evidence that VWF contributes to progression of acute liver injury caused by the hepatotoxin paracetamol.43
VON WILLEBRAND FACTOR REDUCTION: A POTENTIAL NEW THERAPEUTIC OPTION IN LIVER FAILURE SYNDROMES
In view of the emerging evidence of the deleterious effects of raised plasma VWF levels in liver diseases (Table 1), the potential of treatments to reduce VWF needs to be explored in patients with liver diseases. Currently, there are limited data on the role of VWF reduction to treat patients with liver diseases. The beneficial effects of some of the treatments currently in use to treat patients with liver diseases may be due to the VWF-lowering effects of these treatments.
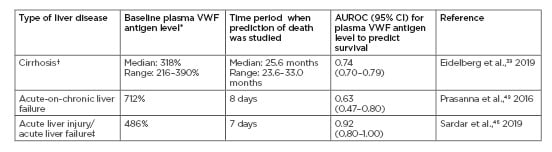
Table 1: Ability of raised plasma von Willebrand factor antigen levels at baseline to predict death in different types of liver diseases in select studies.
(Normal plasma VWF antigen levels are 50–150%).
*Optimal cut-off level of plasma VWF antigen level.
†Meta-analysis of five studies of patients with cirrhosis (N=715 patients).
‡Of the 24 patients with acute hepatotoxicity caused by a single agent (rodenticide), 20 patients had acute liver injury, three had acute liver failure, and one had uncomplicated acute hepatitis.
AUROC: the area under the receiver operating characteristic; CI: confidence interval; VWF: von Willebrand factor.
N-acetylcysteine is used to treat acute liver failure as a result of paracetamol overdose and other causes. Glutathione repletion is a recognised mechanism to explain how N-acetylcysteine exerts its benefits in these patients. N-acetylcysteine has been used for many years to treat chronic obstructive pulmonary disease by reducing levels of mucins (the main protein component of mucus). Chen et al.64 explored whether N-acetylcysteine has a similar effect in reducing VWF multimers, which polymerise in a manner similar to mucin multimers. They found that N-acetylcysteine reduced the size and activity of VWF in human plasma and mice. N-acetylcysteine reduces the disulfide bonds linking VWF dimers.64
Fresh frozen plasma infusions provide ADAMTS13 supplementation, which, in turn, reduces plasma VWF levels (ADAMTS13 causes proteolysis of VWF). Infusions of fresh frozen plasma have been used to treat a patient with noncirrhotic portal hypertension and portopulmonary hypertension, who also had severe ADAMTS13 deficiency.37
Therapeutic plasma exchange dramatically increases survival rate in patients with thrombotic thrombocytopenia purpura (TTP).65 ADAMTS13 deficiency linked to a genetic trait (congenital TTP or Upshaw–Schulman syndrome) or due to anti-ADAMTS13 antibodies (in haemolytic uraemic syndrome after Escherichia coli diarrhoea), is associated with ultra-large-molecular-weight forms of VWF in the circulation. VWF reduction in therapeutic plasma exchange occurs as a result of two mechanisms: the removal of the ultra-large VWF multimers during plasmapheresis, and ADAMTS13 supplementation by infusion of fresh frozen plasma or cryosupernatant from healthy donors to replace the plasma removed from the patient.
Preliminary reports in acute hepatotoxicity (acute liver injury and acute liver failure) because of yellow phosphorus (rodenticide) poisoning suggest that therapeutic plasma exchange is the most potent of these VWF-reducing treatments. In a small number of patients, institution of VWF-reducing treatment, as per a management protocol tailored to the degree of liver dysfunction, improved survival in patients with acute liver injury and acute liver failure, without liver transplantation.46
Fresh frozen plasma contains VWF; however, the question is, is it safe to transfuse or exchange plasma in patients with acute liver injury or failure, who already have raised plasma VWF levels? Plasma exchange appears to improve survival in patients with acute liver failure46,66 and acute liver injury.46 It is likely that in acute liver failure patients, the plasma removed during plasma exchange has high VWF content, and is replaced by plasma obtained from healthy donors (with normal VWF content). Transfusion of fresh frozen plasma to supplement ADAMTS13, as a means to reduce VWF, appeared beneficial in a single patient with ADAMTS13 deficiency and portopulmonary hypertension,37 as well as in acute liver injury.46 Further studies are needed to explore the risks and benefits of plasma exchange and transfusions as a VWF-lowering strategy in patients with liver failure. It is important to obtain blood samples for assays for VWF or for ADAMTS13 prior to initiation of any of these VWF-lowering treatments.
CONCLUSION
Recent reports document raised plasma VWF levels as predictors of poor survival in different syndromes of liver disease. These clinical observations suggest that the markedly raised plasma VWF levels in syndromes of acute liver failure and ACLF may contribute to the pathogenesis of liver failure and multi-organ failure by impeding microcirculatory perfusion in the liver and other affected vital organs. Experimental evidence now confirms that VWF contributes to progression of acute liver injury. VWF-reducing treatments may offer a new therapeutic avenue in liver failure syndromes. Of the different VWF-lowering treatments available, therapeutic plasma exchange appears to be the most efficacious. Current understanding of the dynamics of VWF metabolism in liver failure is rudimentary. There are scarce data on treatments targeting VWF in patients with liver diseases. Further studies are needed to apply these insights to better treatments for patients with liver disease.