Meeting Summary
Prof Peter Ferenci opened the meeting by providing a background to Wilson disease (WD), an enigmatic condition where no two cases are the same. He explored the aetiology, peak age of presentation, and long-term outlook.
Dr Gideon Hirschfield considered the wide variation in WD symptom presentation, the lack of a diagnostic gold standard, and the difficulties around choosing WD endpoints for clinical trials. He went on to consider how study endpoints have evolved over time, and how, in real-life clinical practice, therapies need to be tolerable for patients with negative copper balances.
Prof Anil Dhawan focussed on diagnostic challenges in paediatric WD, reviewing the size of liver biopsies needed for measurement of liver copper dry weight, the penicillamine challenge test, and Leipzig scores. Regarding treatment, he stressed that improvements in liver scores take time on chelation therapy, making it important not to rush patients to transplant. Prof Dhawan explored the development of disease severity scores for transplantation, including the revised cut-off points for the Nazer score. He provided reassuring data around the success of living related liver transplantation from parents heterozygous for WD and raised the possibility of auxiliary liver transplants.
Prof Karl Heinz Weiss considered three WD cases reflecting different aspects of the condition. The neurological case showed deterioration of neurological symptoms after starting D-penicillamine. This, Prof Weiss speculated, may relate to treatment causing shifts in the copper pool from bound copper to unbound copper. The second case involved a young woman with WD who was planning a pregnancy; Prof Weiss showed the importance of patients remaining with therapies they are used to. Finally, he considered a patient with decompensated cirrhosis referred for liver transplantation but for whom, when reassessed with the modified Nazer score, the level did not indicate the need for transplantation. The patient showed side effects with one treatment but subsequently did well on a second treatment and was delisted for transplant.
Introduction
Professor Peter Ferenci
Prof Ferenci explained that many misconceptions exist around WD, including that the condition only occurs in children and young adults, is a neurologic disease, can be excluded if levels of caeruloplasmin are normal, and is a rare condition. Prof Ferenci estimated that in the USA alone, there are at least 9,000 WD patients.
The WD genetic defect affects the copper-transporting adenosine triphosphatase (ATPase) gene (ATP7B) responsible for incorporating copper into copper-binding proteins (including caeruloplasmin) and excreting excess copper into bile. Presentation of WD is highly variable, with no two cases resembling each other. WD patients present with liver disease (including acute hepatitis, fulminant hepatic failure, chronic hepatitis, or cirrhosis), Coomb’s negative haemolytic anaemia, or neurological symptoms (including tremor, rigor, dyskinesia, inability to write, slurred speech, inability to walk, and depression).
The primary defect is hepatic copper accumulation, resulting in liver fibrosis, inflammation, and cell necrosis, which may lead to hepatitis and cirrhosis. Symptoms such as Kayser–Fleischer (KF) rings and central nervous system complications occur only when copper accumulates outside the liver.
An Austrian database involving 1,300 patients, presented by Prof Ferenci, showed the peak age of WD presentation to be between 16 and 20 years.1 However, the oldest patient Prof Ferenci is currently treating was diagnosed when aged 74 years.
Unpublished liver histology data by Prof Ferenci revealed that rates of cirrhosis are low in children but increase slowly towards adulthood (~60% of WD patients have cirrhosis by the age of 20 years). The huge variability, he added, is underlined by findings that some WD patients aged from 60–70 years have not developed cirrhosis.
Although a range of diagnostic tests exist for WD (including caeruloplasmin, urinary copper excretion, and hepatic copper content), no test, except genetic methods, can be considered diagnostic on its own,2 see Figure 1.
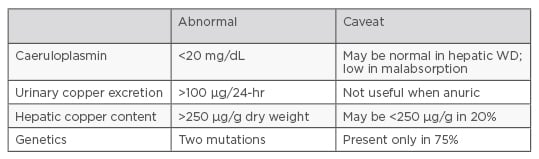
Figure 1: Diagnostic tests for Wilson disease.2
WD: Wilson disease.
Prof Ferenci highlighted the case of a woman diagnosed with WD at the age of 14 years in 1965, who, despite stopping treatment in 1968 (and who has not continued any other subsequent treatment), went on to have two children and showed no clinical evidence of liver damage when examined in 2016.3
A study exploring long-term outcomes in 229 Austrian WD patients diagnosed between 1961 and 2013 showed, at a mean observation period of 14.8 years after diagnosis, that 26% were symptom free, 25% had stabilised, 24% had improved, 17% had deteriorated, and 7% had died.4 Such wide-ranging outcomes, said Prof Ferenci, demonstrate how little is known about the natural history of WD, including disease penetrance.
A problem for assessing treatment efficacy in WD, explained Prof Ferenci, is that the natural history of WD is unknown, and the only parameters are clinical improvement, specific tests focussed towards patients with neurological WD, and copper levels. The ultimate endpoint was survival.
What is the Mark of Success in Treating Wilson Disease Patients
Doctor Gideon Hirschfield
One of the challenges of WD, said Dr Gideon Hirschfield, is the wide spectrum of symptoms that affect the liver, brain, eyes, blood, kidneys, joints, pancreas, heart, and endocrine system.5 See Figure 2.

Figure 2: Different presentations of Wilson disease.5
Such wide variation in WD presentation results in difficulties choosing endpoints for clinical trials, often making it necessary to judge treatment success according to how patients first present with WD. Current treatments for WD, including D-penicillamine, zinc salts, and trientine have been available since the late 1960s.
An additional issue is the lack of a gold standard for WD diagnosis. Diagnosis relies on a ‘composite’ of clinical features and laboratory findings (such as increased urinary copper excretion, reduced levels of serum caeruloplasmin, and high concentrations of liver tissue copper) and KF rings.6,7
Key findings for WD diagnosis include urinary copper levels >100 µg/24-hour, hepatic copper levels >250 µg/g dry weight, caeruloplasmin levels <200 mg/L, and the presence of KF rings. While mutation analysis may provide definitive diagnosis, some variants are not responsible for the disease.
Assessing treatment response therefore is a challenge and WD study endpoints have evolved over time. In 1956, Walshe8 considered removal of copper from the body as a marker of D-penicillamine treatment success but by 1982 was using symptom improvement as a marker of success for trientine in symptomatic neurological WD patients (reviewing factors such as dysarthria and tremor).9 The challenge for the 21st century, said Dr Hirschfield, is to define treatment safety and efficacy more precisely, using clinical scores to determine whether WD symptoms have improved. Current endpoints in clinical trials for WD include quantitative neurologic and speech tests, complete blood cell counts, liver function tests, blood levels (amylase, lipase, creatinine, urea nitrogen, uric acid, and iron variables), and urine protein levels.10
In a recent retrospective analysis comparing outcomes for patients who received D-penicillamine and trientine, Prof Weiss used treatment outcomes including effects on neurologic and hepatic symptoms, and adverse events leading to discontinuation of therapy, but did not include negative copper balances.11 A second paper by Prof Weiss comparing copper chelators and zinc salts used hepatic deterioration (defined as an increase in activity of liver enzymes aspartate aminotransferase [AST], alanine aminotransferase [ALT], and gamma glutamyltransferase) as the study endpoint.12
Dr Hirschfield explained that, when considering real-life clinical practice, therapies need to be tolerable and patients need to achieve negative copper balances, as this effectively stops disease progression.
There was also a need for neurologic stability and improvement. Hepatic stability and improvement can be gauged through clinical means, bloods and imaging, and clinicians should aim where possible to achieve an absence of side effects and demonstrate measurable impacts metabolism. However, Dr Hirschfield explained the field still did not know the exact treatment thresholds clinicians should aim for.
Considering practical issues, Dr Hirschfield said that non-adherence represents a major problem, with studies showing 25–45% of WD patients are non-adherent.13,14 Although direct measurement of free copper (as opposed to protein bound copper) is possible, it is not routinely available and most measurements have yet to be standardised.15-17
While 24-hour urinary copper excretion reflects the fraction of free copper in serum, wide variations have been observed for WD patients. Some recommend stopping chelator treatment for 48 hours, which has low patient acceptance.2
Recommendations suggested by Dr Hirschfield include monitoring efficacy with 24-hour urinary copper excretion. For D-penicillamine and trientine, he suggested complete blood counts to check for cytopenia and urinalysis to check for proteinuria; while for zinc he suggests liver tests for efficacy and creatinine for toxicity.5
During chelation therapies, 24-hour urinary copper excretion outputs of 3–8 μmol per day (200–500 μg) denotes adequate treatment; while for zinc therapy the 24-hour copper excretion target of <2 μmol per day (125 μg) are adequate.18
of <2 µmol per
Important considerations, said Dr Hirschfield, include adherence (once daily treatment may prove more practical), ease of use and monitoring, travel (difficulties occur around treatment refrigeration), availability (different countries have different reimbursements), and follow-up. Treatment follow-up, concluded Dr Hirschfield, needs to be both over the short and long-term and should be appropriate to the severity of WD neurological and hepatic symptoms.
Management of Wilson Disease in the Paediatric Population, Including the Role of Transplantation
Professor Anil Dhawan
WD, said Prof Anil Dhawan, is uncommon in children <3 years of age; it involves mainly hepatic presentation <10 years and neurological symptoms do not usually start until children reach 12–13 years. Disease response changes with age. Unusual WD presentations include haemolytic anaemia, incidental abnormalities of liver enzymes, rickets, gall stones, and neuropsychiatric symptoms. Prof Dhawan described three paediatric WD cases:
- Case 1 involved a 12-year-old girl with a 2-week history of jaundice and haemolytic anaemia, (test scores: caeruloplasmin: 0.1; urine copper: 7 mmol/L: post-D-penicillamine: 26; international normalised ratio (INR): 4; white cell count (WCC): 14; albumin (ALB): 21; AST: 163). The patient had a Wilson index score of 12, which resulted in her undergoing transplantation, after which she has remained well for 10 years. Mutation analysis showed that she was homozygous for common mutations. At birth, her sister was found to be homozygous for the same mutation but to have normal liver function. At 2 years, the sister was started on zinc acetate but at 5 years showed abnormal AST/ALT with liver biopsy revealing steatosis, mild fibrosis, and elevated liver copper. The family opted to change treatment to trientine. Prof Dhawan said that since serum zinc was higher than normal, this raised questions around whether the patient experienced zinc failure or was non-compliant.
- Case 2 involved a 14-year-old girl with decompensated liver disease, ascites, and a WD index of 8. Other centres might have sent her for a transplant. Both ascites and liver function improved over the next 2 years but 3 years later she presented with neurological features resulting in psychiatric hospitalisation. The sad outcome (the girl showed progressive neurological illness and died) raised questions about whether she might have benefited from liver transplant.
- Case 3 involved two siblings. The older boy was diagnosed aged 6 years, after presenting with abdominal pain, and was found to have incidental liver steatosis, to be homozygous for WD, and to have normal liver and neurology. Subsequently, his 3-year-old brother had homozygous mutations identified and showed abnormal liver enzymes (AST: 67; ALT: 71) and on ultrasound was found to have mild splenomegaly.
The diagnosis of WD in children, said Prof Dhawan, includes urine copper, serum copper, serum caeruloplasmin, KF rings, liver copper, Coomb’s negative haemolytic anaemia, biochemical indices, serum zinc, alkaline phosphatase, and molecular diagnostics. Liver copper >250 µg/g of dry weight has become the gold standard for WD diagnosis, with recommendations to use the entire core of the liver biopsy specimen >1 mg dry weight for valid copper content.
A study by Yang et al.19 concluded that liver samples >1 mg dry weight are needed to reflect hepatic copper content. However, Prof Dhawan showed that liver copper could be reliably measured in children when the biopsy sample was <1 mg. This sample size could reliably distinguish WD from autoimmune disease and non-alcoholic liver disease (p<0.001).20 So the message here, said Prof Dhawan, is that one or two decent-sized cores are sufficient.
The penicillamine challenge test, measuring urinary copper excretion for 24-hours after the administration of two doses of D-penicillamine (500 mg), was shown in 1992 to have a sensitivity of 88.2% and specificity of 98.2% for diagnosing WD, using cut-off levels of urinary copper >25 µmol/24-hour.21
When Prof Dhawan re-evaluated the penicillamine challenge test in 38 children diagnosed with WD and 60 controls with other liver disorders, he showed a sensitivity of 76% and a specificity of 93%.22 Furthermore, he found sensitivity was better in symptomatic patients (92%) than asymptomatic patients (46%).
In 2001, a diagnostic score was proposed at the 8th International Meeting on Wilson disease, Leipzig, Germany, taking into account liver copper, rhodanine stain, urinary copper, serum caeruloplasmin, KF rings, neurological features, Coomb’s negative haemolytic anaemia, and genetic ATP7B analysis.23 See Figure 3.
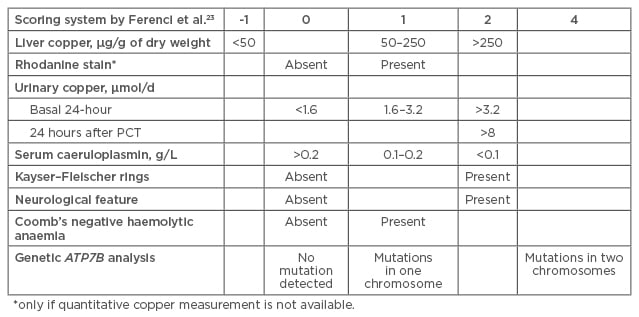
Figure 3: Scoring system for Wilson disease diagnosis.23
The diagnostic score was retrospectively validated in children and shown to have a high specificity (96.6%) and sensitivity (98.1%).
PCT: penicillamine challenge test.
Consensus was reached that scores of ≥4 established WD diagnosis. When Prof Dhawan retrospectively validated the Leipzig score in children he found that it showed high specificity (96.6%) and sensitivity (98.1%).24
With WD treatments for children, clinicians need to appreciate the length of time that may be needed for symptoms to improve. A review of outcomes of 58 children with WD treated at King’s College Hospital, London UK, over 37 years showed that in the 20 children who achieved normal liver function with chelation time to normalisation was 12.2 years for INR, 9 years for AST, and 2.3 years for bilirubin.25
Such delays, stressed Prof Dhawan, underline the importance of not rushing to transplant. Liver transplantation, he said, has its own consequences with children swapping WD treatments for antirejection treatments. The exception was fulminant WD, which without transplantation has nearly 100% mortality.
There is undoubtedly a need to recognise WD features predicting a need for transplant. In 1986, Nazer,26 from King’s College, London, UK, developed a scoring system for the severity of hepatic dysfunction on admission. This was evidenced by prolongation in prothrombin time, AST activity, and serum bilirubin. The study showed patients with scores of ≥7 had higher likelihood of dying or needing a transplant. When Prof Dhawan and colleagues reviewed the data years later (adding WCC and ALB levels) the cut-off was extended to 11.25
In WD paediatric liver transplantation, parents (usually heterozygous for WD) often volunteer as donors leading to concerns around heterozygous donors increasing risk of recurrence. However, results from the Kyoto Experience (involving 11 parent donors donating parts of their liver to children aged 6–16 years) showed marked reduction in urinary copper excretion in all recipients and KF ring improvement.27 The study concluded living related liver transplantation (LRLT) was an excellent choice for effective treatment. The data is further supported by a Chinese study showing survival in WD patients with complicated neurologic manifestations undergoing LRLT.28 Such data, said Prof Dhawan, suggests LRLT ameliorates the neurologic consequences of WD, although the study suggests that outcomes can be related to severity of neurological symptoms at time of transplant. Another possibility is auxiliary liver transplant with Korean doctors reporting a patient who, 26 months after auxiliary liver transplant, had normal serum caeruloplasmin without neurologic problems.29
WD transplant outcomes are reassuring; a study following-up medical records for 121 French WD patients (transplanted between 1985 and 2009) found that actuarial patient survival rates were 87% at 5, 10, and 15 years.30
At King’s College Hospital, increased recognition of psychological issues affecting compliance and quality of life have, in the past 5 years, led to joint WD clinics with hepatologists, neurologists, and psychologists. While expensive, said Prof Dhawan, treatment adherence and patient quality of life have greatly benefited.
How to Manage Difficult Situations when Dealing with Wilson Disease Patients
Professor Karl Heinz Weiss
Prof Karl Heinz Weiss considered the lessons that could be learnt from 3 WD cases: a symptomatic neurologic patient, a patient who wished to become pregnant, and a decompensated cirrhosis patient. The cases allowed him to explore a range of considerations around WD treatment.
The symptomatic neurologic case was a 21-year-old male who after referral for tremor was found to have elevated transaminases, caeruloplasmin 0.03 g/L (normal range 0.2–0.6 g/L), urinary copper excretion >2 times the upper limit of normal (ULN), and was found to be KF positive. After WD diagnosis, the patient was started on D-penicillamine (20 mg/kg bw). He had barely any evidence of hepatic disease but the tremor was severe. Prof Weiss said it was important to use a neurologic rating scale in the clinic for neurologic symptomatic patients, as it helps quantify the neurologic symptom burden and follow the clinical course of the disease. Following 1 week of therapy, this patient was unable to walk or swallow and his urinary copper excretion increased dramatically (>35 x ULN).
One of the risk factors for neuro deterioration reported in the literature has been starting on too high a dose of D-penicillamine or trientine.31-33 A possible explanation, suggested Prof Weiss, is that chelating agents may be responsible for shifting the copper pool, i.e. moving copper from the CP-bound copper (non-toxic pool) to the non CP-bound toxic pool (toxic pool) and shifting too much copper into the circulation leading to neurological worsening.34 When Litwin35 used the Unified Wilson Disease Score scale on 143 symptomatic WD patients, diagnosed between 2005 and 2009, he found neurological worsening following treatment initiation in 11.1% of patients. But the role of free copper remains unclear as it was not assessed in that paper.
The current patient underwent D-penicillamine dose reduction and achieved slow stabilisation over the next 8 weeks but showed no improvement making it necessary to consider other treatment options. In the Heidelberg Vienna cohort study, a retrospective analysis of 288 WD cases for hepatic treatment failure, Prof Weiss and Prof Ferenci showed that hepatic treatment failure occurred least often with chelating agents (p<0.001) and actuarial survival was greater for chelating agents (p<0.001).12 On this basis, the patient was prescribed a combination treatment with trientine and zinc. The advantage of a combination is that drugs have different modes of action, but the disadvantage is that the regimen is complicated as both drugs need to be taken at widely spaced intervals to avoid interference between zinc and chelator ions.36,37 After 6 months, the patient showed neurologic recompensation. In treating symptomatic neurological disease, said Prof Weiss, the rule should always be to start low and go slow.
The second patient, a 25-year-old woman with no neurologic symptoms diagnosed with WD at the age of 14 years after mild transaminase elevation, said that she hoped to become pregnant. Ultrasound showed no cirrhosis, no ascites, and mild splenomegaly. Transaminases, ALB, INR, bilirubin, and haemoglobin were within normal range.
As long as a woman’s liver is healthy enough to become pregnant, hepatic worsening does not appear to be a frequent finding. A study by Pfeiffenberger et al.38 reviewing pregnant women showed that hepatic worsening is a rare event for all treatments.38
Considering the possibility of switching treatments, Prof Weiss said it might take 6–12 months for the transaminases to increase in cases who were non-responders to one treatment.12 By far the easiest approach, said Prof Weiss, would be to advise the patient to stay with the same treatment but to decrease the dose.
Decompensated cirrhosis. The third patient was a 22-year-old male diagnosed with WD after referral for ascites. He had elevated transaminases, caeruloplasmin 0.05 g/L, urinary copper excretion >2 x ULN, KF positive, INR 1.9, ALB 26 g/L and was listed for transplant. However, on the Modified Nazer score he had a score of 6, which is well below the transplant ‘cut-off’ of 11.25 Over 6 months of treatment the patient showed slow recompensation and was de-listed for transplant.
Medical therapy, said Prof Weiss, should always be indicated even for very sick patients with subacute liver failure. The overall prognosis for WD patients if they are adherent and on drugs is excellent. A study by Bruha et al.39 that followed 117 WD patients showed that their long-term survival did not differ from the general Czech population (p=0.95).39
Discussion
The wide-ranging audience questions considered the value of liver biopsies, doses, genetic testing, and issues around pregnancy. Regarding liver biopsies, Prof Ferenci said he had reviewed data on 100 patients with established WD and found 18% did not have cut-off levels for copper. Other studies have found that copper is unevenly distributed in the liver and a biopsy may not accurately assess its level. Urinary copper analysis was complementary to biopsy, he said.
The challenge of genetics, said Dr Hirschfield, is that people with the same genotype do not necessarily have the same phenotypes. Someone with significant illness can have a sibling with the same mutation who is completely asymptomatic. Prof Ferenci stated that when patients are diagnosed with WD it should be mandatory to offer genetic testing to first degree relatives.
With regard to chelator dose, Prof Weiss said that, if adult patients are doing well with normal liver function tests and there is no residual neurologic disease, clinicians should aim to lower the dose. Dr Hirschfield agreed that there was likely to be a point where less copper chelation was needed to keep the patient in clinical equipoise, adding that dose reduction had benefits for side effects and cost. Prof Dhawan explained that because paediatric patients are growing, unchanged doses are equivalent to a reduction.
With regard to female WD patients contemplating pregnancy, Prof Ferenci said that the likelihood of their partners carrying the gene was low. Partners should not be considered for genetic testing but once born the child should be tested to confirm that it is heterozygote.
In pregnancy, the risk of an untreated mother dying far outweighs any potential teratogenic effects of therapy. Also, added Prof Ferenci, women with WD who do not take therapy have high risks of miscarriage. Prof Weiss said that in pregnancy the best choice for most women is to remain on existing treatments they have been previously stable on.