Presenters: Ronald Sokol,1,2 Tamir Miloh,³ Noelle Ebel,⁴ Ryan Himes⁵
1. Pediatrics, Children’s Hospital Colorado, USA
2. Pediatrics-Gastroenterology, Hepatology and Nutrition, University of Colorado School of Medicine, USA
3. Pediatric Transplant Hepatology, Miami Transplant Institute, Florida, USA
4. Pediatric Gastroenterology, Stanford University, California, USA
5. Pediatric Hepatology, Ochsner Hospital for Children, Jefferson, Louisiana, USA
Disclosure: Sokol has served as a consultant for Albireo Pharma, Mirum Pharmaceuticals, and Alexion Pharmaceuticals. Miloh has served as a consultant and received speaker fees from Mirum Pharmaceuticals. Ebel has served as a consultant and has received speaker fees from Mirum Pharmaceuticals. Himes has served as a consultant and received speaker fees and research funding from Albireo Pharma, Mirum Pharmaceuticals, Alexion Pharmaceuticals, and Travere Therapeutics.
Acknowledgements: Writing assistance for this independently written article was provided by Gemma Glasscock, Health Interactions, London, UK.
Disclaimer: This summary was written independently of the live presentations, and the presenters did not provide input on this article.
Support: The symposium, poster, and the publication of this article were funded by Mirum Pharmaceuticals, Inc.
Citation: EMJ Hepatol. 2022;10[Suppl 2]:2-11.
Summary
Cholestatic liver diseases, caused by impairment in bile formation or disruption of bile flow in the liver, are the leading indications for paediatric liver transplant, and result in significant morbidity and mortality.1-3 Rare, chronic, cholestatic diseases that present in childhood include Alagille syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC), and biliary atresia (BA). The differential diagnosis of these diseases is broad, with clinical manifestation varying according to aetiology; however, early biochemical evidence of cholestasis includes elevations of liver transaminases, and total and direct bilirubin.4
Management of BA requires early diagnosis and surgical intervention, while for patients with ALGS and PFIC, the treatment goals are to relieve the leading symptom, cholestatic pruritus, and to also improve nutritional status, correct vitamin deficiencies, and manage the complications of advanced liver disease such as ascites and variceal bleeding.3,5 Pruritus is one of the most bothersome symptoms of cholestatic liver disease in children and, until recently, no approved pharmacological treatments were available, with off-label agents such as ursodeoxycholic acid, bile acid sequestrants, and rifampicin being the mainstay of treatment.5 However, many children do not respond to these approaches, and invariably require surgical alternatives and liver transplant. The recent approval of ileal bile acid transporter (IBAT) inhibitors have provided a much-needed option for pruritus relief for children with ALGS and PFIC.6,7 This article summarises a poster at the American Association for the Study of Liver Diseases (AASLD) and a symposium at the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN), which increase understanding of these rare diseases and discuss new pharmacological approaches to treat them.
Predictors of 6-Year Event-Free Survival in Patients with Alagille Syndrome Treated with Maralixibat
Ronald Sokol
At the 2021 AASLD Liver Meeting®, Sokol examined predictors of long-term event-free survival (EFS) in children with ALGS. This analysis included 76 maralixibat-treated participants with ALGS from three long-term clinical trials for up to 6 years, who were followed for the development of their first clinically significant event. A clinically significant event was described as liver transplant, surgical biliary diversion, hepatic decompensation (ascites requiring therapy and variceal bleeding), or death. Participants who were on maralixibat for 48 weeks from the first dose, and had lab results at 48 weeks, were included. The results of this analysis demonstrated that variables predictive of EFS included: total bilirubin at Week 48, serum bile acid (sBA) level at Week 48, pruritus change from baseline to Week 48 (assessed by caregivers using the Itch-Reported Outcome [Observer] [ItchRO(Obs)] scale), and age at initiation of maralixibat (Figure 1). Kaplan–Meier analyses demonstrated that at Week 48 lower total bilirubin levels (<6.5 mg/dL versus ≥6.5 mg/dL) and lower sBA levels (<200 μmol/L versus ≥200 μmol/L) were associated with increased EFS (90% versus 43%; p<0.0001, and 85% versus 49%; p=0.0010, respectively). A larger reduction in ItchRO(Obs) scores from baseline to Week 48 (>1 point reduction versus ≤1 point reduction) and a higher age at initiation of maralixibat were also associated with improved EFS (88% versus 57%; p=0.0046, and 83% versus 57%; p=0.0059, respectively). Sokol explained that since pruritus is often an indication for liver transplant in children with ALGS, these data demonstrate that resolution of pruritus following the use of maralixibat is significantly associated with improved EFS and transplant-free survival (TFS).
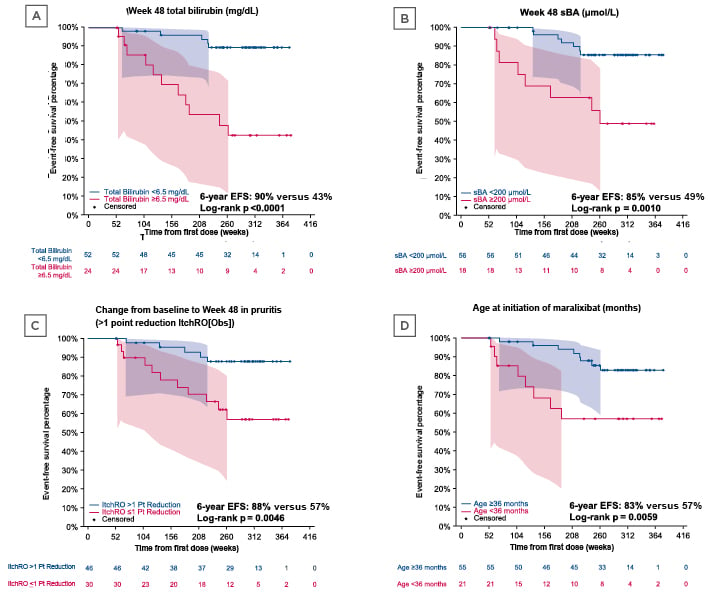
Figure 1: Event-free survival according to A) total bilirubin at Week 48, B) serum bile acid level at Week 48, C) pruritus change from baseline to Week 48, and D) age at initiation of maralixibat.28
Data values under each panel indicate the number of patients at each time point. Analysis examined predictors of long-term EFS, including TFS, in patients with ALGS enrolled in three clinical trials of maralixibat, with up to 6 years of follow-up; included patients who were on maralixibat 48 weeks from the first dose, and had lab results at 48 weeks in the analysis.
ALGS: Alagille syndrome; EFS: event-free survival; ItchRO(Obs): Itch-Reported Outcome (Observer); pt: point; sBA: serum bile acid; TFS: transplant-free survival.
Introduction to Paediatric Cholestatic Liver Diseases
Tamir Miloh
Cholestatic liver diseases are caused by impairment in bile formation or disruption of bile flow in the liver and, as Miloh explained, are typically characterised by jaundice and pruritus. Intractable pruritus is a frequent and often debilitating symptom that is difficult to manage, causing sleep disturbances and impaired quality of life.8,9 There is an overlap of clinical symptoms in ALGS, PFIC, and BA, which makes early diagnosis challenging at times.10-12 Even if a correct and timely diagnosis is made, the majority of children will require a liver transplant by adulthood; data from the ChiLDReN Network study showed a TFS probability rate of 24% in a cohort of 293 patients with ALGS,13 and data from the multicentre Global ALagille Alliance (GALA) Study Group demonstrated that in 911 participants with ALGS, only 41% reached adulthood with their native liver.14 With poor TFS across ALGS, PFIC, and BA, there is a clear need to develop alternative treatment options.13-17
Miloh emphasised that the first step towards diagnosis usually begins with prolonged jaundice (>14 days) beyond the early neonatal period, which results in the infant being tested for cholestasis.11,18,19 Laboratory work, with further analysis of λ-glutamyltransferase levels, can help to differentiate between ALGS and PFIC subtypes.20,21 In recent years, the availability of comprehensive genetic testing has significantly improved diagnosis of these rare cholestatic diseases.22 In the majority of institutions, liver biopsies are still performed to ensure timely and accurate diagnosis of BA; however, distinguishing BA from ALGS is critical, as patients with BA require a Kasai procedure <60–90 days after birth, while patients with ALGS who are misdiagnosed with BA and receive a Kasai have worse outcomes.11,13-15 Once a diagnosis is confirmed, the key treatment goals for managing children with ALGS, PFIC, and BA are to provide symptomatic pruritus control, and to improve growth and development.11,13,15,19
The enterohepatic circulation of bile acids serves as a feedback mechanism to inhibit bile acid synthesis and maintain bile acid homeostasis. Disrupted bile flow from the hepatocyte to the biliary canaliculi leads to accumulation of bile acids in hepatic cells, causing cellular dysfunction, necrosis, inflammation, apoptosis, and alterations in liver function and,if left untreated, eventually leads to end-stage liver disease.3,23-25 To reduce this bile acid accumulation, surgical or pharmacological approaches can be used to block the recirculation of bile acids to the liver.3
In the NAtural course and Prognosis of PFIC and Effect of biliary Diversion consortium (NAPPED) study, surgical biliary diversion in children with PFIC2 controlled sBA, and was associated with a significantly increased probability of TFS.15 Due to its key role in bile acid reuptake, IBAT is a rational target for pharmacological interruption of bile acid recirculation, and studies with IBAT inhibitors (such as maralixibat [Mirum Pharmaceuticals, Inc., Foster City, California, USA] and odevixibat [Albireo Pharmaceuticals, Inc., Boston, Massachusetts, USA]) in ALGS and PFIC have demonstrated significant improvements in pruritus, and reductions in sBA.6,7,26 Maralixibat has also demonstrated an improvement in the probability of TFS in patients with ALGS and PFIC2.27-29 Maralixibat was approved in 2021 for the treatment of cholestatic pruritus in patients with ALGS who are 1 year of age and older.6 Odevixibat was also approved in 2021 for the treatment of pruritus in patients with progressive familial cholestasis who are 3 months of age and older.7
Constructing a New Approach in the Management of Alagille Syndrome
Noelle Ebel
ALGS is a rare, life-threatening, multisystem disease that presents in childhood with a range of clinical manifestations, and occurs due to mutations in two genes associated with the Notch signalling pathway: JAG1 (occurring in 89–94% of cases) and NOTCH2 (in up to 4% of cases).13,30,31 These mutations can result in a variety of clinical manifestations, including chronic cholestasis, characteristic facial features (high prominent forehead, pointed chin, and deep-set eyes), ophthalmic abnormalities, cardiovascular disease, skeletal abnormalities, renal dysfunction, and vascular dysfunction. Diagnosis is based on the presence of three out of these seven main organ systems, when family history or molecular diagnosis is not available.32 ALGS has a reported incidence of 1 in 30,000–50,000; however, this is likely an underestimation given incomplete penetrance, and the highly variable phenotype that can frequently be mistaken for other diseases.33
The cholestatic pruritus associated with ALGS is often referred to as the most severe of any liver disease, negatively impacting quality of life, and is a leading cause of liver transplantation in these patients.3 Data from the multicentre GALA Study Group showed that only 41% of 911 participants with ALGS reached adulthood with their native liver;14 similar patterns were seen in the ChiLDReN Network study, which showed a TFS probability rate of only 24%.13 Those who receive a liver transplant can benefit from improvements in growth and resolution of pruritus;34-36 however, despite liver transplantation being an effective therapy for a range of chronic liver diseases in children, it can result in a number of potentially fatal or long-term complications such as graft rejection and failure, hypertension, and chronic renal impairment, with a 5-year mortality rate of 11.6%.37,38 Post-transplant immunosuppression increases the risk of infections and malignant complications;39-41 it can result in significant burden for children and their caregivers, particularly when accounting for the prolonged post-transplant survival of children, who require immunosuppression throughout their lives.42,43
Ebel went on to discuss the utilisation of maralixibat in the Phase IIb ICONIC trial, a study with a 4-week double-blind, placebo-controlled, randomised drug withdrawal period (RWD) in participants with ALGS.44 The study enrolled 31 children, and maralixibat was administered according to a dose escalation schedule over 6 weeks of treatment to a final dose of 380 μg/kg, in which participants were able to increase to twice-daily dosing after Week 100.26
The primary endpoint was mean change in fasting sBA levels from Weeks 18–22, and pruritus manifestation was explored as a secondary endpoint, assessed using observer- (ItchRO[Obs]), patient- (ItchRO[Pt]), and clinician-rated (Clinician Scratch Scale [CSS]) scales.26 Ebel demonstrated that significant and sustained improvements were seen in pruritus following maralixibat treatment, with 84% achieving a clinically meaningful decrease in ItchRO scores (≥1 point reduction during the 48-week period; Figure 2). The impact of maralixibat was highlighted during the RWD from Week 18 to Week 22. After significant reductions in sBA from baseline to Week 18 (-88 µmol/L) for both the placebo (n=16) and maralixibat groups (n=13), the maralixibat group maintained this significant sBA reduction during the RWD, whilst the placebo group had a return of sBA levels close to baseline.26 When maralixibat treatment was resumed following the end of the RWD, decreases in pruritus measures and sBA levels were maintained long-term until Week 204.26 Further investigation demonstrated that reductions in pruritus intensity significantly correlated with reductions in sBA levels (r=0.47; p=0.012), as well as reductions in CSS scores following maralixibat treatment.26,45
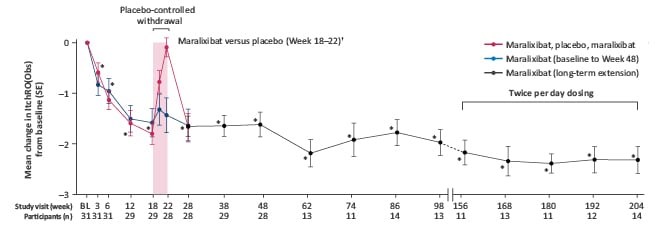
Figure 2: Change in pruritus (ItchRO[Obs]) from baseline to Week 204 in patients in the ICONIC study.26
Dashed lines represent data not shown between Week 98 to Week 156. Twelve participants went to twice per day dosing on the basis of raised sBA in the open-label extension.
*95% CI excludes zero (compared with baseline, overall population).
†The maralixibat, placebo, maralixibat treatment group (n=16) received placebo during the randomised withdrawal period (purple-shaded area), whereas the maralixibat treatment group (n=13) continued to receive maralixibat.
Reprinted from Gonzales E et al.,26 with permission from Elsevier.
BL: baseline; CI: confidence interval; ItchRO(Obs): Itch-Reported Outcome (Observer); sBA: serum bile acid; SE: standard error.
Ebel noted that maralixibat treatment was well-tolerated throughout the study period,26 with most adverse events (AEs) being transient in nature and mild-to-moderate in severity, with no deaths reported. The majority of gastrointestinal AEs occurred within the first 4 weeks of treatment and lasted less than 1 week in duration.26 Diarrhoea and abdominal pain were the most frequent AEs, and occurred with a similar incidence between the maralixibat- and placebo-treatment groups during the RWD (8% for both diarrhoea and abdominal pain in maralixibat-treated participants; 6% for both for placebo-treated participants).26 The expected side effects of IBAT inhibitors include disturbances that are gastrointestinal in nature and, as such, the gastrointestinal-related AEs observed are considered in line with the drug’s mechanism of action.26
Odevixibat, another IBAT inhibitor, has also been investigated as a treatment for ALGS. The Phase II, open-label, dose-finding study in children (n=24) with pruritus due to ALGS (n=6), PFIC (n=13), BA (n=3), or other intrahepatic cholestasis causes (n=2), showed improvements in sleep and pruritus scores, and reductions in sBA levels from baseline.46 Generally, odevixibat was well-tolerated in the study population, and displayed similar gastrointestinal disturbances as expected of the drug class.46 The ongoing Phase III ASSERT study will further explore the use of odevixibat in ALGS.47
Ebel gave a summary of a 2021 AASLD late-breaking oral presentation from the GALA study group, who recently presented data on children with ALGS, comparing time to first clinical event in maralixibat-treated participants (n=84) versus an historical control cohort (n=469).27 A clinical event was defined as biliary diversion surgery, a decompensation event (ascites requiring therapy or variceal bleeding), liver transplantation, or death.27 Selection criteria were pre-specified to ensure cohorts were aligned as closely as possible, with both baseline and disease characteristics being well-balanced between cohorts.27 Ebel explained that children treated with maralixibat had a significant improvement in EFS at 6 years, compared with the GALA control cohort, showing a 70% reduction for clinical outcomes with maralixibat treatment versus the natural history of patients with ALGS (p<0.0001). This was confirmed consistently across multiple sensitivity and subgroup analyses to strengthen the robustness of the findings.27
Ebel summarised her presentation to recap the challenges associated with the management of ALGS, and the severe impact cholestatic pruritus can have on the patient’s quality of life. The ICONIC trial resulted in significant and sustained improvements in pruritus, leading to the recent approval of maralixibat for the treatment of cholestatic pruritus in patients with ALGS 1 year of age and older.6,26
Establishing New Foundations for Children with Progressive Familial Intrahepatic Cholestasis
Ryan Himes
PFIC is a heterogeneous group of diseases classified into six subtypes, which all disrupt bile formation,48-50 and has an estimated incidence of 1 in 50,000–100,000 births, of which PFIC2 accounts for nearly half of all cases.48,51 PFIC2 is the most aggressive of the subtypes, with the clinical severity linked to the type of ABCB11 mutation, resulting in a varied phenotypic spectrum, ranging from jaundice to pruritus and portal hypertension, hepatomegaly, scleral icterus, and failure to thrive.15,19,52-54 Himes illustrated the traditional course of PFIC2 through a case study presentation, before moving on to discuss recent advances with IBAT inhibitors. Progress has been seen with maralixibat and the recently approved IBAT inhibitor odevixibat, which was studied in the PEDFIC 1 Phase III study and ongoing PEDFIC 2 open-label extension study. Odevixibat was administered at 40 or 120 μg/kg/day versus placebo in children with various PFIC subtypes, and data have shown significantly positive pruritus assessments in those receiving odevixibat (n=42) versus those treated with placebo (n=20; p=0.004). In addition, significantly more participants achieved an sBA response when treated with odevixibat than placebo (p=0.003).55,56 Overall, odevixibat was well-tolerated and there were no deaths during the study, with only one participant discontinuing due to diarrhoea.55
Maralixibat, another IBAT inhibitor, was studied in the open-label, Phase II INDIGO study in 33 paediatric participants with PFIC1 or PFIC2.57 Participants were administered maralixibat 266 μg/kg daily, which increased to 266 μg/kg twice-daily. Maralixibat resulted in a reduction of sBA levels with long-term treatment, improvements in cholestatic pruritus (as measured by ItchRO[Obs]) and improvements in growth.29 Five-year TFS was seen in participants with sBA control (n=7) following maralixibat treatment, who all experienced normalisation of liver parameters throughout this period (Figure 3).29 Throughout the INDIGO study, maralixibat was generally well-tolerated, with the most frequently reported AEs being nasopharyngitis, vomiting, cough, and diarrhoea.29 All participants with PFIC1 or PFIC2 experienced a treatment-emergent AE, with 78.9% deemed potentially maralixibat-related and 15.8% leading to discontinuation. Importantly, no deaths occurred during the study period.29 Maralixibat is now being studied at higher doses and in other PFIC subtypes, and these outcomes will be reported from the Phase III MARCH-PFIC study, which is currently enrolling.
Figure 3: Five-year transplant-free survival in participants with serum bile acid control following maralixibat treatment.29
Treatment responders were defined as the seven participants with PFIC2 who had complete resolution or clinically relevant reduction of pruritus with maralixibat treatment. Clinically relevant reduction in pruritus is defined by improvement by ≥1 point from baseline in the ItchRO score.
ItchRO: Itch-Reported Outcome; PFIC: progressive familial intrahepatic cholestasis.
Structuring New Outcomes for Biliary Atresis
Tamir Miloh
BA is a rare cholestatic liver disease in infancy, occurring in approximately 1 in 12,000 live births in the USA, 1 in 18,000 in Europe, and 1 in 5,000 in Japan.11,58 BA presents with progressive cholangiopathy and patients rapidly develop progressive fibrosis and cirrhosis if left untreated.59,60 The cause of BA is unknown; however, factors implicated in the aetiology and pathology are thought to occur during development or shortly after delivery, and include injury by viruses, toxins, or inflammation or, to a lesser extent, genetic mutations.59,60 BA has overlapping features with other cholestatic diseases; for accurate diagnosis, histopathology of resected material and intraoperative cholangiogram are performed.18 Prompt diagnosis is critical, and performing Kasai procedures early (before 44 days) is associated with an improved TFS.61 Lower levels of total serum bilirubin at 3 months post-Kasai are also indicative of a higher probability of TFS.62 Data suggest that sBA levels can be predictive of a successful response to a Kasai procedure: in participants with an sBA level ≤40 µmol/L, 2.3% of children experienced death or had a liver transplant by 144 months, whereas in children with sBA levels >40 µmol/L, this proportion increased to 32.3% (p=0.0006).63 These data led to the introduction of bilirubin neonatal screening programmes, which in the USA has resulted in the Kasai procedure being performed at an earlier average age (36 days versus 56 days).63,64
There are currently no approved pharmacological treatments for BA, with no standard therapeutic approach post-Kasai. In the START study, initiating steroids after the Kasai procedure did not decrease TFS, but was associated with earlier onset serious AEs.65 Miloh indicated that there is an unmet need for therapies that could reduce the need for liver transplant, and discussed the potential role of IBAT inhibitors. Studies are ongoing to assess whether IBAT inhibitors can improve outcomes in BA, specifically to reduce sBA levels, improve nutrition, and reduce the rate of fibrosis progression, pruritus, and other extrahepatic complications associated with end-stage liver disease.17,26,66 Odevixibat is being investigated in 200 participants with BA post-Kasai compared with placebo in the Phase III BOLD study for 2 years, with the primary endpoint of TFS.67 Similarly, maralixibat is being investigated in 72 children with BA in the Phase II EMBARK study, with a 26 week endpoint using bilirubin and sBA levels. Participants can extend treatment out to 2 years.68
Conclusion
ALGS, PFIC, and BA are rare, developmental, multisystem diseases that typically present with chronic cholestasis and debilitating pruritus. Clinical studies of pharmacological IBAT inhibition have demonstrated promising results in children with ALGS and PFIC. As a result of these approaches, two new IBAT inhibitors have been approved recently for these cholestatic liver diseases. Maralixibat is indicated for the treatment of cholestatic pruritus in patients with ALGS who are 1 year of age and older, whilst odevixibat is indicated for the treatment of pruritus in patients with PFIC who are 3 months of age and older. Recent data have shown that lower sBA, total bilirubin, and ItchRO(Obs) scores are all predictive of improved EFS and can be used as a marker for long-term outcomes, providing a rationale for pursuing IBAT inhibition as a promising alternative to surgical intervention. Further data on these exciting new approaches to the management of cholestatic liver disease in children are eagerly awaited.
Question and Answer Session
The panel ended the symposium with a question and answer discussion segment, with the first topic addressing odevixibat dosing, and the question: “Reduction in sBAs seemed less dramatic at the higher dose of odevixibat; is that correct, and what is the hypothesised mechanism?” Himes explained that despite appearing numerically less, statistically the drug was compared to placebo and, therefore, the reduction at both doses was likely similar.
Another question asked: “Given that IBAT inhibitors are reversible, do you expect improved suppression of bile acid reabsorption with pre-meal dosing three times daily?” Miloh responded that it depends on the half-life of the medication and increasing the dose does not necessarily provide a better outcome. Himes added that it is recommended to take IBAT inhibitors pre-meal whether the drug is administered once- or twice-daily, and that there is sufficient itch relief with once- or twice-daily dosing, rather than three times daily. Miloh added that it is possible that combining IBAT inhibitors may improve outcomes, although there are no trials ongoing to investigate this.
Ebel went on to discuss how IBAT inhibitors could fit into clinical practice, particularly in light of recent data demonstrating an improvement in EFS. Miloh commented that reducing sBA levels appears to reduce progression of liver disease compared with standard anti-pruritic medications, and it will be interesting to see how these IBAT inhibitors affect other cholestatic processes. Ebel highlighted the importance of ensuring patient advocacy groups and healthcare practitioners are educated on these rare diseases, for example, through scientific seminars, and how valuable this is for families. Himes and Miloh echoed this, and added their own experiences, noting that preparedness is key to productive conversations with families and patients. Ebel made a final point regarding measurement of sBA and, with the advent of IBAT inhibitors, the importance of testing. Miloh commented that with medications now being available to reduce sBA levels, measuring sBA should be standard of care.