
Abstract
Migraine is a chronic and disabling disorder affecting >1 billion individuals worldwide.
Current treatments for the prevention of migraine include antihypertensives, antiepileptics, and antidepressants, and all share limited tolerability and adherence, highlighting the need for the development of new disease-specific and mechanism-based agents. In this context, four novel anti-calcitonin gene-related peptide monoclonal antibodies have been investigated in a large Phase II–III clinical programme and showed similar efficacy to the currently used drugs for migraine prevention but with a significantly improved safety profile, as highlighted in this review. It is expected that patient compliance with treatment will increase with the use of these therapies, improving the long-term overall outcome of migraine. However, real-world evidence is needed to confirm the tolerability and safety of anti-calcitonin gene-related peptide monoclonal antibodies before the drugs can be established as first-line agents in the prophylactic treatment of migraine.
INTRODUCTION
Migraine is a pervasive brain disorder that is ranked as the second most disabling condition1-3 and has the third highest prevalence among all medical illnesses.4 The common understanding that has guided the management of people with migraine is pharmacological intervention to prevent disease chronification.5 Pharmacological agents approved for the prevention of episodic migraine (EM)6 span different drug classes (e.g., antihypertensive compounds, tricyclic antidepressants, and antiepileptic drugs).7 Due to the prolonged administration required for migraine prevention and drug nonspecificity, these drugs can cause numerous adverse events (AE), and the agents interact with many other medications in comorbid patients.8 Furthermore, only one medication (onabotulinumtoxinA) is approved for the prevention of chronic migraine (CM).9-15 The suboptimal efficacy and tolerability of current treatments contribute to poor patient treatment compliance and adherence (up to 68% of patients stopped using preventative medication within 6 months).16-18 As patients cycle through preventative therapies, discontinuation rates increase and the augmented need for abortive medication leads to disease chronification.19 All of the above, in addition to pharmacophobia (fear of medication) and the nocebo effect (experience of AE related to patients’ negative expectations that a treatment will most likely cause harm instead of improving disease),20 suggest the need for novel treatments that are better tolerated and have fewer contraindications, not only for patients who have failed existing preventative treatments but also for treatment naïve patients, especially those who fluctuate in the prechronic phase.6,21,22 Among the available molecular targets, calcitonin gene-related peptide (CGRP) has the best base of evidence for controlling migraine.23 CGRP, a 37 amino acid long neurotransmitter, is part of the calcitonin family of peptides, together with calcitonin, amylin, and adrenomedullin, and is the most potent microvascular dilator currently known.24-26 CGRP acts on an unusual receptor family that is located at sites crucial to the triggering of migraine, including the cerebrovasculature, the trigeminocervical complex in the brainstem, and the trigeminal ganglion (Table 1).24,27,28 Small molecule CGRP receptor antagonists, the gepants, are under development for the treatment of migraine, with three (rimegepant, ubrogepant, and atogepant) such agents currently in a Phase III clinical trial programme.29-36
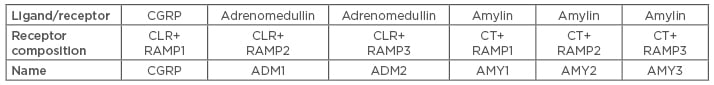
Table 1: The calcitonin gene-related peptide receptor family.
CGRP binds to both CGRP and AMY1 receptors.24
ANTIMIGRAINE MONOCLONAL ANTIBODIES
Monoclonal antibodies (mAb) against CGRP share several pharmacokinetic advantages over small anti-CGRP molecules (e.g., greater target specificity and prolonged half-life, making them suitable for monthly administration to prevent migraine). Three of these macromolecules target the CGRP ligand (fremanezumab, galcanezumab, and eptinezumab), while a fourth (erenumab) targets the CGRP receptor.14,37,38 All four require parenteral administration and have a preferential peripheral site of action, since only 0.1–0.5% of the mAb cross the blood–brain barrier due to their large size (molecular weight around 150 kDa).14,39,40 These four mAb have shown particular effectiveness for the prevention of both EM and CM.41,42
Erenumab
Erenumab (AMG 334) is a fully human IgG2 mAb that prevents native CGRP ligand binding to the CGRP receptor.6 At 70 mg, the estimated elimination half-life of erenumab is 21 days, supporting monthly subcutaneous dosing and, thus, bettering patient compliance.43,44 Even in the early phases, studies showed no significant differences among healthy subjects and patients with migraine in least squares mean 24-hour or nocturnal diastolic blood pressure (BP) measurements between placebo and erenumab-treated groups.6 Similar results were found from studying the coadministration of erenumab and sumatriptan and their effect on resting BP in healthy subjects (no additional effect on resting BP beyond the effects of sumatriptan monotherapy, without affecting the pharmacokinetic of sumatriptan).45 The AE that were most commonly reported in the single-dose study (in ≥20% of subjects in the erenumab group) were headache in healthy subjects (erenumab: 25.0%; placebo: 25.0%) and nasopharyngitis (erenumab: 50.0%; placebo: 50.0%), arthralgia (erenumab: 33.3%; placebo: 0%), and influenza-like illness (erenumab: 33.3%; placebo: 16.7%) in patients with migraine. No deaths or serious AE (SAE) were reported in the Phase I studies. Most AE were mild or moderate in severity and there were no clinically meaningful changes in laboratory assessments or vital signs.6
Again, in Phase II clinical trials for prevention of both EM and CM, the number of patients with AE was similar between the treatment groups. In EM, 95% of the patients in the erenumab treatment groups experienced AE that were mild or moderate in severity (similar in all different doses) versus 98% in the placebo group. The most common AE was nasopharyngitis, and the reported SAE were considered to be unrelated to treatment. A small percentage of participants developed binding and neutralising anti-erenumab antibodies, with no apparent association recorded among these patients in terms of AE, safety, or efficacy. The incidence of injection-site reactions was low (5%) and all reactions were mild in severity. No notable findings were recorded following the collection of clinical and laboratory results, vital signs, BP, or ECG changes. One death was noted: a 52-year-old man with a history of migraine with aura, and this was confounded by pre-existing cardiovascular risk factors (3-year history of diagnosed hypertension, obesity, a lipoprotein level of 153 mg/dL, left anterior hemiblock on baseline ECG, and a family history of myocardial infarction). The patient’s autopsy showed evidence of severe coronary atherosclerosis and the presence of cardiac stimulants (phenylpropanolamine and norpseudoephedrine) in the liver. The myocardial ischaemia event was based on results of an exercise treadmill test, which showed transient exercise-induced myocardial ischaemia, confounded by sumatriptan administration 4 hours prior to the event. It was considered not related to treatment by the investigator.17,44
In studies of CM, the most frequent AE, reported by ≥2% of erenumab-treated patients, were injection-site pain, upper respiratory tract infection, nausea, nasopharyngitis, constipation, muscle spasms, and migraine.46 Taking into consideration the individuality of patients and the nocebo effect, with the common denominator being the AE associated with prior medication failure, the incidence of AE was broadly comparable within each group (placebo and different doses of erenumab).15
Notably, a placebo-controlled study of erenumab in a high-risk population of patients with stable angina with a median age of 65 years showed that intravenous erenumab 140 mg did not lead to significant changes in exercise time compared to placebo. The change in treadmill exercise time from baseline was noninferior for erenumab compared to placebo, no difference was observed in the time to onset of ≥1 mm ST-segment depression or exercise-induced angina, and there were no significant differences between treatment groups in reported AE through the 12-week safety follow-up.47
The most common AE reported in Phase III clinical trials are fatigue, nasopharyngitis and upper respiratory tract infection, injection-site pain, headache, vertigo, and nausea.14 Once more, the development of anti-erenumab binding and transient neutralising antibodies was observed, but with no clinical or other association. No clinically meaningful differences between the erenumab groups and the placebo group were observed with regard to the results of hepatic function testing, creatinine levels, total neutrophil counts, vital signs, or ECG findings.14,42,48,49
Fremanezumab
Fremanezumab (TEV 48125) is a fully humanised IgG2a/kappa mAb that potently and selectively binds to both α and β isoforms of CGRP.50 The mean half-life values range from 32–36 days.51 In early clinical studies, treatment-related AE occurred in 21.2% of subjects receiving fremanezumab (no association pattern regarding the dosage), compared with 17.7% in those receiving the placebo. The most common treatment-emerging AE reported were headache, nasopharyngitis, gastroenteritis, and back pain, while the two SAE, thoracic aortic aneurysm (patient had an unreported history of Ehlers–Danlos syndrome) and glaucoma, in individuals receiving fremanezumab, were not treatment related. Fremanezumab was also not associated with any clinically relevant patterns of change in vital signs (including systolic and diastolic BP, temperature, and heart rate), ECG parameters, or laboratory findings.50 Similar results were reported in Phase I studies comparing the prevalence of safety issues between Caucasian and Japanese healthy subjects.51
In Phase II clinical trials of both CM and high frequency EM,52,53 despite having different doses, the AE (treatment-emerging and treatment-related) reported had consistency regarding the type and frequency. In the CM Phase II study,53 40% of patients in the placebo group, 53% in the 675/225 mg dose group (675 mg in the first treatment cycle and 225 mg in the second and third treatment cycles), and 47% in the 900 mg dose group had treatment emergent AE. In the Phase II trial of high frequency EM, the rates of treatment-emergent AE were 56% (placebo), 46% (225 mg fremanezumab), and 59% (675 mg fremanezumab).52 The most common AE in both studies were mild injection-site pain and pruritus. The SAE that occurred were not treatment-related and, again, there were no relevant changes in BP or other vital signs. The prevalence of patients with detectable concentrations of fremanezumab antibodies (1% in both studies) was much lower than that detected with other monoclonal anti-CGRP antibodies (19% with galcanezumab and 14% with eptinezumab).52,53 In a post-hoc analysis of the previous studies, it was found that fremanezumab was compatible with most of the major classes of migraine preventative therapies, which suggests that it will be a useful and safe agent as an add-on therapy for patients requiring additional preventative treatment. Also, results suggest that fremanezumab can be started immediately, without requiring other preventatives to be titrated or washed out first, giving patients the opportunity for a more rapid clinical improvement.54
In Phase III studies regarding CM, AE were reported in 64% of the patients receiving placebo, 70% of those receiving fremanezumab quarterly, and 71% of those receiving fremanezumab monthly; the reported AE were mild-to-moderate in severity in 95–96% of patients in all three groups. Again, the most common AE were injection-site reactions (40% placebo, 47% fremanezumab quarterly, and 47% fremanezumab monthly), the severity of which did not differ significantly among the trial groups. SAE occurred in 2% of the patients given placebo, 1% of those given fremanezumab monthly, and <1% of those given fremanezumab quarterly, and no participants had anaphylaxis or a severe hypersensitivity reaction. Abnormalities in hepatic function occurred in 1% of patients in the fremanezumab groups and <1% in the placebo group, which can be attributed to the use of nonsteroidal anti-inflammatory drugs. As in previous studies, antidrug antibodies developed in two patients who received fremanezumab quarterly. No clinically significant changes in vital signs, physical examination findings, or ECG results occurred in any of the trial groups.55
In a Phase III study of EM,56 66%, 66%, and 58% of patients who received fremanezumab monthly, fremanezumab once (higher dose), and placebo, respectively, reported at least one AE. Treatment-related AE were higher in the fremanezumab groups (48% in the monthly group and 47% in the single-higher-dose group) compared with placebo (37%), with the most common being injection-site reactions (pain, induration, and erythema). No relevant changes in vital signs (BP, pulse, temperature, and respiratory rate), physical examination measurements (including weight), or ECG findings were noted in patients in any of the treatment groups. There were no clinically significant changes in any laboratory parameters, including liver function tests. Again, a small percentage of patients in the fremanezumab monthly dosing group developed antidrug antibodies against fremanezumab, without any significant AE. It should be noted that one patient died 109 days after receiving a single higher dose of fremanezumab. The patient had withdrawn from the study 38 days earlier because of a family emergency and the cause of death noted in the autopsy report was suicide by diphenhydramine overdose; this death was considered unrelated to the treatment.56
Galcanezumab
Galcanezumab (LY 2951742) is a humanised mAb with a long half-life (time to maximum serum concentration ranges from 7–13 days and elimination half-life is about 28 days) that binds to both α and β CGRP isoforms with approximately equal affinity.57 AE reported in a Phase I clinical trial were transient, with no apparent relationship with the prolonged systemic drug exposure (indicated by the long half-life of galcanezumab). In subjects receiving galcanezumab, the most common AE were headache, nasopharyngitis, haematuria, and contact dermatitis; the frequencies of these AE were similar to placebo. Other frequently reported AE in subjects receiving galcanezumab were diarrhoea, toothache, and increased alanine aminotransferase. There were no apparent differences among galcanezumab dose groups or between galcanezumab dose groups and placebo in terms of frequency of any AE. This observation included changes from baseline in vital signs, laboratory values, and ECG parameters. It was reported that 26% of the galcanezumab-treated subjects produced antidrug antibodies, the presence of which had no obvious effect on pharmacokinetics and pharmacodynamics compared with subjects who had no detectable antidrug antibody titres.4
In a Phase IIa study58 (galcanezumab 150 mg administrated subcutaneously twice a month), AE were reported by 72% of patients in the galcanezumab group and by 67% in the placebo group (no significant difference). AE that occurred more frequently in galcanezumab versus placebo included injection-site pain, erythema, or both (21 [20%] of 107 patients versus 7 [6%] of 110 patients), upper respiratory tract infections (18 [17%] versus 10 [9%]), and abdominal pain (6 [6%] versus 3 [3%]). There were two SAE reported in the treatment arm and four in the placebo arm, none of which were deemed to be related to the study drug. Once more, there were no clinically important changes in laboratory parameters, ECG results, or vital signs between the groups. Antidrug antibodies were detected in 8 patients at screening and in 20 patients at the end of the study; nevertheless, there was no association in terms of efficacy and AE with the antidrug antibodies.58
In a Phase IIb study (galcanezumab 120 mg once per month),59 a similar frequency of AE was reported in both the placebo (70 [51.1%]) and galcanezumab-treated (140 [53.1%]) patients. The most common AE for galcanezumab were injection-site pain, which had a dose-dependent response, upper respiratory tract infections, nasopharyngitis, dysmenorrhoea, and nausea, without any dosage correlation; most AE were mild-to-moderate in intensity. None of the SAE were considered to be related to galcanezumab.59 Taking into account the vital signs during treatment and post-treatment periods, mean changes in systolic BP, diastolic BP, and pulse were not clinically meaningful, and there were no trends to show that galcanezumab treatment increased BP. Also, mean baseline-to-endpoint changes in ECG intervals (PR, QRS, and QTcF) and heart rate showed no clinically meaningful differences between individual or pooled galcanezumab dose groups and placebo. In addition, changes in temperature were small and not clinically meaningful; weight changes were also similar across treatment groups.59,60
Larger studies (Phase III EVOLVE-161 and EVOLVE-218) have corroborated the aforementioned findings. In EVOLVE-1,61 5 participants in the placebo group and 6 in the galcanezumab 120 mg group reported a total of 12 SAE, none of which were considered by the investigator to be associated with the treatment. Similarly, in EVOLVE-2,18 the percentages of SAE, which were 1.1%, 2.2%, and 3.1% for the placebo, galcanezumab 120 mg, and galcanezumab 240 mg groups, respectively, did not differ significantly. Injection-site erythema, injection-site pruritus, and injection-site reactions were the most frequently reported AE related to the injection site for galcanezumab compared with placebo in both Phase III clinical trials, but most AE were mild-to-moderate in severity. Discontinuations owing to AE in galcanezumab-treated patients were low (2.2–4.0%). The most common post-treatment emergent AE was upper respiratory tract infection, which occurred at a similar rate across treatment groups. Other post-treatment emergent AE that occurred in ≥1% of patients in the combined galcanezumab group were viral upper respiratory tract infection, sinusitis, and influenza, and these events occurred at a rate similar to placebo. Again, there were no statistically significant differences between galcanezumab dose groups and placebo on mean change from baseline of systolic BP and pulse at any visit. For temperature, statistically significant mean increases (≤17.6°C) were observed only in EVOLVE-1, and these were transient and not sustained. Body weight was measured at Month 6 only and the mean change from baseline to last observation carried forward endpoint was small (<1 kg) and not statistically significant between treatment groups. Regarding the development of anti-galcanezumab antibodies, at baseline, in EVOLVE-1, 5.9% of patients in the placebo group (n=25) and 8.9% (n=18; 120 mg group) and 10.8% (n=23; 240 mg group) in the galcanezumab dose groups had antidrug antibodies present. With consistency, the respective percentages in EVOLVE 2 were 8.4% (placebo), 8.1% (galcanezumab 120 mg), and 11.2% (galcanezumab 240 mg). The percentage of patients with antidrug antibodies during the double-blind treatment phase was low, and the number of patients with neutralising antibodies was even less. No antidrug antibodies were associated with changes in efficacy or safety.18,61
Eptinezumab
Eptinezumab (ALD403) is a humanised anti-CGRP IgG1 antibody that potently and selectively binds to both the α and β forms of human CGRP. The plasma half-life of eptinezumab after an intravenous infusion of 1,000 mg is 31 days. In a Phase II clinical trial,62 during which patients with frequent EM were given one intravenous dose of 1,000 mg of eptinezumab, AE were experienced by 52% of patients in the placebo group and 57% in the eptinezumab group. The most frequent AE in both groups were upper respiratory tract infection, urinary tract infection, fatigue, back pain, nausea, vomiting, and arthralgia. During the study, 55% of patients experienced ≥1 AE. No infusion reactions were reported during the study and most AE were transient and mild-to-moderate in severity. Six SAE were reported by three patients; all of these events were deemed to be unrelated to the study drug (fractured fibula, pyelonephritis, non-cardiac chest pain, and transient ischaemic attack). There were no clinically significant differences in vital signs, 12-lead ECG results, or laboratory safety data between patients treated with eptinezumab or placebo at any time during the study. Furthermore, in antidrug antibody assays, 14% of patients in the eptinezumab group who were tested had positive results, suggesting the potential formation of eptinezumab antibodies during the study. However, the corresponding antidrug titres were low and no obvious effects of immunogenicity on the pharmacokinetic parameters or efficacy were noted.62
In all succeeding clinical trials, Phase II and III for CM and PROMISE 1 and 2, the observed safety profile of eptinezumab was similar to placebo. PROMISE 163 is a double-blind, randomised, placebo-controlled Phase III study evaluating the efficacy and safety of eptinezumab in patients with frequent EM. Both the safety profile and the placebo rates were consistent with previously reported eptinezumab studies.63,64 On the other hand, the Phase III trial, PROMISE 2,65 is a evaluating the safety and efficacy of eptinezumab for CM prevention. As previously mentioned, AE rates among eptinezumab-treated subjects were similar to placebo-treated subjects. Likewise, the most commonly reported AE for eptinezumab, occurring at an incidence of ≥2%, were nasopharyngitis (6.3%), upper respiratory infection (4.0%), nausea (3.4%) and urinary tract infection (3.1%), arthralgia (2.3%), dizziness (2.6%), anxiety (2.0%), and fatigue (2%).65
CONCLUSION
Monoclonal antibodies against CGRP and its receptor have passed all clinical phases and are becoming available in the USA and Europe. Unlike formerly available prophylactic treatments for migraine, anti-CGRP mAb have been developed specifically for the prophylaxis of migraine following a mechanism-based design. Their profile regarding dosage, pharmacokinetics, and distribution makes anti-CGRP mAb attractive in terms of adherence and patient compliance. Many safety questions were raised due to preclinical data that came from studying and blocking CGRP,66 but no safety flags occurred during the large programme of their development and all four CGRP mAb have shown similar tolerability and safety in Phase II and III trials. The most common adverse events are reported in Table 2. No interactions with other preventative drugs have been reported. The major scepticism regarding their use was related to the potential cardiovascular effects and liver toxicity. While existing data do not confirm any cardiovascular effect, animal studies and long-term Phase IV trials are needed to further evaluate the safety of anti-CGRP mAb. As far as liver toxicity is concerned, mAb elimination is mainly the result of proteolysis and does not involve metabolism by liver enzymes, making drug–drug interactions and hepatotoxicity unlikely. Thus, the overall safety profile of anti-CGRP mAb for the prevention of migraine has been reported to be more than satisfactory so far.
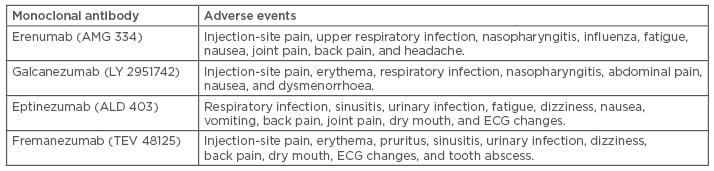
Table 2: The most common adverse events that have been reported more frequently in the active anti-calcitonin gene-related peptide monoclonal antibody arms versus placebo arms.6,14,18,40,41,42,44-49,52-60





