
Abstract
Post-market monitoring of medical devices by manufacturers and regulatory agencies aids the identification of novel hazards or increasing trends in the risks associated with devices. This narrative review estimates the rates of under-reporting of medical device adverse events and explores the reasons and possible solutions. Incident reports may be presented to the manufacturer or the regulatory agency spontaneously by consumers, patients, clinicians, or distributors of medical devices. However, it is evident that reporting does not occur to a great extent, with the rate of reporting estimated to be as low as 0.5% of all occurrences. The programmes and processes to increase and support the reporting of adverse events need to be reviewed, with consideration given to the cost-benefit of increased reporting in relation to the regulator, regulated entities, healthcare facilities, and professionals, as well as the public.
INTRODUCTION
Historically, medical devices have not undergone pre-market clinical trials in the same way as pharmaceutical products. Moreover, medical devices undergo changes and modifications over time that are not subjected to human trials. Therefore, there is a need for strong post-market vigilance of medical devices1 as an integral component of the manufacturer’s quality management system. Vigilance aids in identifying new or escalating risks with a device, as well as possible improvements to the useability or functionality of the device.2 The reporting of device failures to manufacturers or regulators helps identify risks at the earliest possible timepoint. The hazard may be novel or due to regional factors, such as environmental conditions or clinical practice, which were unknown or not initially considered by the manufacturer. Currently, under many jurisdictions, it is compulsory for manufacturers to report adverse events associated with the use of a medical device to the regional regulatory agency that result in, or have the potential to result in, death or serious injury; however, this is not a requirement of healthcare professionals or facilities. Globally, regulators encourage reporting of incidents relating to medical devices by consumers, patients, clinicians, and distributors of devices, but the rate of reporting remains low. For example, the U.S. Food and Drug Administration (FDA) estimates the reporting rate as 0.5%.3 This narrative review will estimate the rates of under-reporting of medical device adverse events and explore the reasons behind this and the possible solutions.
Published qualitative data provide insights into the reasons for the under-reporting of incidents in the healthcare setting; these include a culture of non-reporting in the profession,4 lack of recognition that the incident was related to a medical device,5 or, as is often noted in the spontaneous reports received by Australian regulator, the Therapeutic Goods Administration (TGA), from users of the device, the reporter contacts the TGA only after the expiration of the warranty period when the manufacturer or supplier were providing replacements for faulty items.
ESTIMATING THE RATE OF UNDER-REPORTING
In 2017, of the 5,379 medical device incident reports received by the TGA, 771 (14%) were from sources other than the manufacturer’s Australian legal representative (the sponsor). Sources of spontaneous reports include those from the patient or caregiver, health professionals, and departments within healthcare facilities (Figure 1).
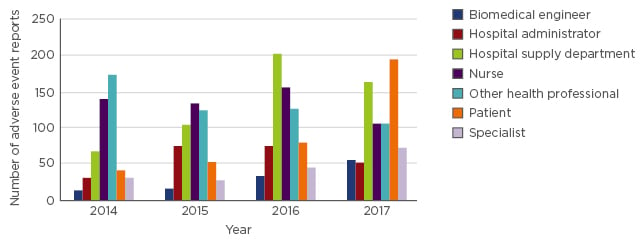
Figure 1: Adverse events spontaneously reported to the Therapeutic Goods Administration (TGA).
The number of adverse events reported to the Therapeutic Goods Administration (TGA) from sources other than the manufacturer’s legal representative is low. The source of the report is shown to change over time, which may be due to education of reporting processes or workplace culture.
Case Study 1: Ventilators
To gain an insight into the prevalence of adverse event under-reporting, data relating to medical devices that are closely monitored in the healthcare setting, such as life-support devices, are optimal for investigation. The authors chose ventilators for closer analysis because they are closely monitored, easy to count, have a life-support function, and are currently the subject of a post-market review,6 meaning additional baseline data are available to the TGA.
To complement the TGA data, additional data were derived from a large public health service,7 chosen because it has a diverse range of services with rural and large urban centres, and also a population profile representative of the entire Australian population. The public health service data showed, on average, one device failure per year for each of the 200 ventilators in service.
These failures were either repaired in-house (66%), repaired under contract service (29%), or repaired under warranty (5%), and none were reported to the TGA. This showed that medical device failures may not be reported to the regulator or the manufacturer when the device is repaired in-house. Alternatively, the manufacturer may consider device replacement as non-reportable, or consider unreturned devices with faults as ‘unconfirmed’.
In Australia, there are approximately 4,170 ventilators that are currently in use in the intensive care setting. If the rate of failure of ventilators (one failure for each ventilator in service per year) is comparable across all healthcare facilities, the number of reports expected by the TGA, just for ventilator failures, would be around 4,000 per annum. However, over the past few years, the TGA has received an average of 15 reports per annum (Figure 2). This can be interpreted to mean that <0.4% of ventilator failures result in a report to the TGA. This study has not been able to determine the proportion of all failures that resulted, or may have resulted, in patient harm; however, as ventilators are considered life-support equipment, any swap-out during use or reduced service availability may have a serious impact on the patient.
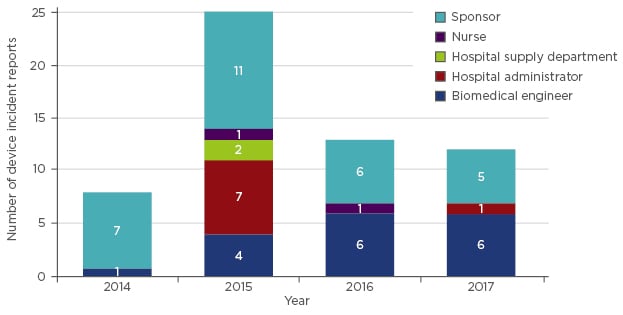
Figure 2: Reports of ventilator-related adverse events in Australia.
Adverse event reports regarding ventilators received by the Therapeutic Goods Administration (TGA), demarcated by source of report.
When looking more broadly to extrapolate the number of patients that may have been impacted from ventilator failures, data from the Australian Institute of Health and Welfare (AIHW)8 show, on average, a patient will be ventilated for 5.5 days. With approximately 4,170 ventilators in use, assuming these are in constant use and they are used on one patient every 5.5 days, the annual number of ventilated patients in Australia is estimated to be 276,000. This means only one report is received by the TGA for every 18,400 patients treated. Combining the two sets of data suggests the number of failures on a per-patient basis is closer to 1 in 66 patients treated; however, the true rate of under-reporting may be much higher since the rate-of-use errors and close calls may be as high as 10–15% of all ventilator user tasks performed.9
Case Study 2: Urogynaecological Mesh
Urogynaecological mesh can be used as another example when estimating the level of under-reporting of adverse events. In Australia, the first adverse event relating to urogynaecological mesh was received by the TGA in 2006, despite the first device being approved for supply in 1998. Until 2012, the TGA received 63 reports of urogynaecological mesh-associated adverse events.10 The increase in public awareness of the complications associated with this kind of device correlated with an escalation in the number of adverse event reports in the following 5 years, with the TGA receiving an additional 186 reports as of 29th May 2017;10 12% of reports were from healthcare professionals, 38% from the sponsor of the device, and 50% from patients. During the Australian Inquiry by the Senate titled ‘Number of Australian women who have had transvaginal mesh implants and related matters’,11 the Health Issues Centre reported that, as of 3rd August 2017, 2,400 women had reported personal accounts of adverse events to their centre.12 Although not all of these reports received by the Health Issues Centre may be unique reports or associated with devices implanted in Australia, there is a clear indication of under-reporting of adverse events with this kind of device.
The literature reports the risk of serious injury for this type of device is ˜10% (ranging from no adverse events reported to 30%).13-17 If this rate is then applied to the supply numbers of the device or patient healthcare records, the number of affected patients could be extrapolated. As of 29th May 2017, in Australia, 151,000 urogynaecological mesh devices have been supplied since 1998,10 and the TGA has received 249 reports of serious adverse events, which equates to a reporting rate of serious adverse events of 1.5%. However, consideration does need to be given to the fact that multiple mesh devices may be implanted into a single patient and not all devices may have been used following supply. While there are multiple variables that are not able to be accurately defined with current data collection strategies, this type of analysis highlights the low incidence of adverse event reporting.
UNDER-REPORTING: REASONS, PROBLEMS, AND POSSIBLE SOLUTIONS
Non-Reporting Culture
The culture of non-reporting by health professionals is multifactorial, including reasons such as fear of blame, lack of time, perceived ineffectiveness of reporting, complexity of reporting, and lack of knowledge of the reporting system. Moreover, if the adverse event is a known complication, can be resolved clinically, or the device failure can be fixed by the practitioner or biomedical engineer, the issue may not be deemed necessary to report.4,18,19
Regulatory agencies have recognised the importance of retrieving this missing data with a variety of initiatives introduced to overcome these issues, including data mining and education programmes. For example, the FDA introduced the Medical Device Safety Network (MedSun) and the Sentinel initiative. The latter aims to use electronic health records, primarily from insurance claims, to extract information relating to adverse events; however, this has had greater success with complications related to pharmaceuticals rather than medical devices due to the lack of data specific to medical devices.3
In Australia, the InSite programme20 was piloted by the TGA in two healthcare facilities in 2014 and 2015, in which an education programme was used highlighting the benefits of adverse event reporting, how to report, and what an adverse event may look like. The participants’ knowledge of reporting was assessed by a qualitative questionnaire and the number of adverse event reports before and after the training was evaluated for each of the facilities. The questionnaire revealed that staff believed the reporting of patient harm in the hospital reporting system would result in the TGA being informed of the incident, and there was a lack of knowledge that the TGA would want to be given this type of information. Regarding the number of reports received by the TGA, there was a 10-fold increase in the number of adverse event reports from one of the sites during the time period that the education programme was conducted. However, when reporting data were analysed the year after the programme was completed and training was no longer provided, the reporting of adverse events returned to the pre-education programme state, thereby confirming that continued education and close interaction and collaboration are required to maintain high levels of reporting of adverse events. In addition, it was evident that the commitment of the administrators of the healthcare facility was a key factor in the success of the programme for each site, with no increase in adverse events reported when there was little support of the programme.
Further to the lack of awareness of the role of the regulatory agency regarding medical devices by healthcare professionals and the public, the ease of reporting is also a factor for under-reporting. There is a lack of alignment of adverse event reporting programmes in healthcare facilities and sponsor’s/supplier’s/manufacturer’s quality management systems with the regulatory agencies’ databases.21,22 For example, in a hospital setting, an adverse event involving a medical device may be reported in the hospital system, but this system does not allow the information to be pushed directly to the regulator. Moreover, the healthcare professional may assume that the regulator has been advised through the hospital reporting system. In addition, fewer reports will be made when there is an obligation to provide detailed reports, especially when reporters have a lack of time or are already over-burdened by administrative tasks.
Non-Association of an Incident with the Medical Device
The failure to recognise the association of a medical device with poor or adverse patient outcomes can be attributed to multiple factors, including the emergence of evidence of a novel complication, extended time periods between the intervention with the medical device and the onset of symptoms, non-specific or seemingly non-related patient signs and symptoms, a lack of education or available information relating to known complications, and a lack of knowledge of the previous intervention due to a different healthcare professional undertaking the procedure.
Novel issues may emerge with the advent of new clinical tests or when a complication has a delayed onset that was beyond the timeframe of any previous clinical trials or retrospective studies. The identification of emerging or novel complications is only possible when the manufacturer or regulatory agency receives adverse event reports. Ideally, the regulatory agency would have the advantage of early identification of complications with a certain kind of device because they would receive reports of devices from multiple manufacturers, enabling an issue to be extracted from the data when it may not be evident from a single model of a medical device.
Case Study 3: Breast Implant-Associated Anaplastic Large Cell Lymphoma
The emergence of breast implant associated-anaplastic large cell lymphoma (BIA-ALCL) is an example of a new complication, first identified in 199723 and following the introduction of breast implants by the Dow Corning Corporation in 1962.24 The awareness and research undertaken into this disease has led to increased identification and reporting of cases over the past 6 years (Figure 3). As of 19th July 2018, the TGA has received information on 75 confirmed cases of BIA-ALCL since 2007. Factors that have influenced the delayed identification of BIA-ALCL as a risk associated with the implantation of the device include the late onset of the disease, low rate of occurrence, and identification of disease markers. The average time of onset of BIA-ALCL is 8 years,25 which exceeds the clinical trial duration for many studies of the implants. Another factor is the low rate of occurrence (1 in 1,000–10,000), which requires a greater number of patients for detection than have previously been enrolled in studies.26 Cytological markers for this disease, namely the presence of CD30+ and the absence of anaplastic lymphoma kinase (ALK), have only recently been identified and are now recommended as clinical determinants for the presence or absence of the disease.27 However, testing for these markers is not routine in all countries, with the cost being too great for many patients. This complication highlights the importance of post-market surveillance of devices and continual analysis of risks associated with the device.
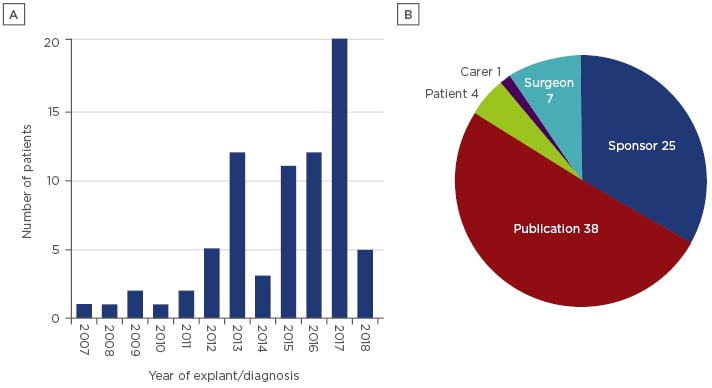
Figure 3: Number (A) and source (B) of reports of breast implant associated-anaplastic large cell lymphoma received by the Therapeutic Goods Administration (TGA).
The reports included are all confirmed as breast implant associated-anaplastic large cell lymphoma, with cytology results indicating CD30+ and ALK-. The source of the reports is predominately publications.
Case Study 2: Urogynaecological Mesh
The failure to recognise or acknowledge that an adverse event has been caused by a medical device, along with the failure to report the event to the regulatory agency or the manufacturer, has been recently demonstrated with the clinical issues arising from the use of urogynaecological mesh. As stated previously, as of 29th May 2017, the TGA had received 249 adverse event reports for this kind of device, but during the Australian Senate Inquiry, the Health Issues Centre reported that, as of 3rd August 2017, 2,400 women had reported personal accounts of adverse events to their centre.12 The large disparity between the number of reports that regulatory bodies receive and the number of patients actually affected is undoubtedly a factor in the delay of action being taken to prevent further harm to patients. In this case, the lack of reporting of adverse events to regulators and manufacturers was not only evident with the specialists who had inserted the mesh but also with the general practitioners, non-implanting specialists, and pain specialists who were treating these women several years after the device was implanted. This may be attributed to the lack of understanding of the potential complications of the device or simply the lack of knowledge of the reporting of adverse events; regulatory agencies rarely advertise their roles associated with medical device failures. A confounding factor in this case is also the number of patients who did not report their debilitating outcomes. This may have been due to this same lack of knowledge of the reporting system, or the perception that the reporting system was too complex and required information that they did not have. Moreover, the lack of reporting may have been a result of the continued dismissal of their symptoms by healthcare professionals, lack of awareness that a mesh device had been implanted, or simply not wanting to discuss such personal complications with a stranger.
There have been similar under-reporting issues highlighted by inquiries into hip and breast implants.28,29 A study into hospitalisations for heart disease after metal-on-metal hip replacements using data from the Australian Orthopaedic Association National Joint Replacement Registry (AOANJRR) and veteran medical records found that, due to the low number of cases reported, a definitive causation was not possible, potentially leading to unnecessary action.30 Additionally, some of the issues highlighted in these inquiries related to known complications that are not generally reported by health professionals because the assumption is that the rate of these complications is identified and acceptable.
To address the lack of reporting by health professionals, many have suggested that registries for implanted devices or mandatory reporting are needed.28,29 However, registries are expensive, take considerable resources to develop and run, and usable data are not generated for several years. Mandatory reporting of deaths and serious injuries from USA health facilities to the FDA31 is one method of attempting to stimulate a higher rate of reporting of the most serious events and, along with other programmes such as Medsun,32 may in turn lead to increased reporting of all events.
ADVERSE EVENT REPORTS
When analysing adverse event reports, not only are the number of reports important but consideration also needs to be given to the type of device; different classes of device will have different levels of post-market vigilance. For example, the sponsors of active implantable medical devices are required to provide annual reports of adverse events and complaints for the first 3 years of inclusion on the Australian Register of Therapeutic Goods, but this is not required for Class I devices. Reports can also be received regarding safety, quality, and performance of a device, and the vigilance and response to each kind of event may be different. In addition, reports may also have varying degrees of information depending on the source, nature, or severity of the report. Moreover, reports might relate to the different stages of the lifecycle of the device, from design through to manufacture, shipping, storage, and use of the device, as well as reprocessing aspects or disposal. The inadequacies in current reporting frameworks may also be a hindrance to recognising the kind of issue and or assisting in appropriate responses to reports.
CONCLUSION
It is difficult to accurately determine the rate of under-reporting of medical device adverse events because the actual number of incidents is unknown. However, the case studies in this review are in agreement with the FDA estimate of a reporting rate of 0.5%. The need for reporting adverse events associated with medical devices is of importance to facilitate the prevention of further harm and to aid in continual design improvement of devices, and, therefore, regulators and manufacturers need to reconsider current methods used to encourage the reporting of events. The recognition of this need for improved vigilance and post-market surveillance has been highlighted in the recent changes to the European Union Medical Device Regulation,33 which includes the establishment of a European database on medical devices. Conversely, consideration also needs to be given to how regulators manage the large amount of reports already being received, even with the strong evidence of significant under-reporting. Regulators are likely to need new and appropriate ways to analyse the data, both in the near and long-term future when, if current reporting trends continue, there will be ever-increasing numbers of adverse event reports.






