Abstract
A quadricuspid pulmonary valve (QPV) is a rare congenital cardiac anomaly, mostly identified incidentally during autopsy or imaging examination. The authors report on an autopsy case report describing a case of a QPV recognised incidentally during the autopsy of a 91-year-old male with bilateral pneumonia. All four cusps of the pulmonary valve exhibited approximately equal sizes and showed no degenerative changes, while the remaining heart valves displayed no structural anomalies. There was no clinical or pathological evidence of stenosis or regurgitation. In this report, the authors discuss the clinical presentation, review the existing literature, investigate the diagnostic and clinical implications of QPV, and highlight the key factors involved in QPV embryogenesis.
Key Points
1. Quadricuspid pulmonary valve is a rare congenital cardiac anomaly, typically clinically quiescent and asymptomatic, mostly identified incidentally during autopsy or imaging examinations.2. The identification of quadricuspid pulmonary valve in living patients is infrequent and often overlooked due to limitations in transthoracic echocardiography imaging. Diagnosis relies on imaging studies, with transoesophageal echocardiogram, cardiac CT, and cardiac MRI being the most sensitive methods, providing superior visualisation of the pulmonary valve.
3. Identifying anatomical variants can significantly impact clinical outcomes by reducing diagnostic delays.
INTRODUCTION
Quadricuspid pulmonary valve (QPV) is a rare congenital cardiac anomaly in which the pulmonary valve has four cusps, typically of various sizes, rather than three symmetrical cusps. QPV is usually an asymptomatic, isolated malformation that is difficult to detect clinically using routine strategies such as transthoracic echocardiography (TTE), and is thus under-diagnosed. Historically, most cases have been discovered incidentally during post-mortem examination, but recent advances in cardiac imaging technologies and frequent health examinations have resulted in more incidental diagnoses of QPV. The precise embryology of QPV is unknown, but may result from a developmental anomaly during embryonic cardiac outflow tract septation and valvulogenesis.
CASE PRESENTATION
A 91-year-old male, fully recovered from multiple surgeries following various cancer diagnoses, was admitted for hypoxemic respiratory failure with a low-grade fever and tachycardia. Imaging revealed consolidation in the lower left lung and small bowel obstruction. The patient also had sinus tachycardia with occasional ventricular tachycardia, Escherichia coli infection, and low white blood cell counts. Despite antibiotic treatment and respiratory support, the patient’s blood pressure, heart rate, and respiratory rate fluctuated over the next 24 hours, and he passed away the following day. The autopsy confirmed extensive bilateral pneumonia as the cause of death.
The authors examined the heart thoroughly and identified certain abnormalities. The heart was slightly enlarged, weighing 456 grams. There was evidence of moderate coronary artery disease, with up to 25% stenosis of the left anterior descending and right coronary arteries, and 50% stenosis of the left main coronary artery. The heart showed mild left ventricular hypertrophy. No evidence of acute or chronic myocardial infarction, endocarditis, valvular stenosis, or regurgitation was identified. When examining the valves, the team identified a QPV consisting of four approximately equal-sized cusps, none with significant degenerative changes (Figure 1). The adjacent pulmonary wall was unremarkable, with normal diameter and thickness. The thickness of each valve was 2 mm, with no fenestrations, calcifications, or fibrosis. Additionally, the aortic valve displayed mild nodular calcifications and fibrous thickening, predominantly affecting the lower-to-mid cusps. Given the lack of associated clinicopathologic findings with the QPV, it was felt to be an incidental finding.
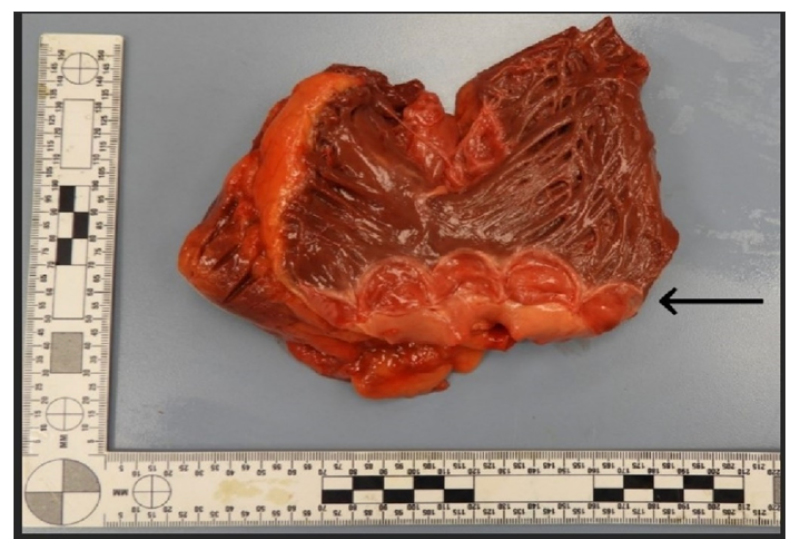
Figure 1: Autopsy heart with opened right ventricle and root of pulmonary artery.
A quadricuspid pulmonary valve with no gross pathologic change is present (arrow).
DISCUSSION
QPV is a rare congenital cardiac malformation with an incidence from 0.1–0.2% at autopsy, and 0.2% in a European study of donor hearts.1,2 It is more frequent in males, with a male-to-female ratio of 2:1.1 Advancements in cardiac imaging techniques, regular health screenings, and cardiac surgery have led to an increased number of incidentally discovered QPV.3,4 However, it is challenging to determine the true incidence of QPV given the retrospective nature of these autopsy studies, and in addition, QPV may be frequently overlooked given its clinically silent nature.5
The classification of QPV can be done using the Hurwitz and Roberts system, which categorises the valves based on the relative size of the supernumerary cusp in relation to the remaining cusps.1 Type B, the most common variant, accounts for 60% of cases, and features three cusps of equal size with a smaller fourth cusp. Type A represents 12% of cases and is characterised by four cusps of similar size, such as seen in the present case. Type C represents 15% of cases and exhibits two larger cusps with two smaller cusps. The remaining cases belong to Types D–G, which features four cusps of different sizes.1 An exploration of embryology is helpful for understanding the basis of these variants.
The semilunar valves develop from endocardial cushions located at the atrioventricular junction and in the cardiac outflow tract (OFT) during the 5th week of gestation.6 The septation of the OFT plays an integral role in the development of semilunar valves. Initially, the embryonic cardiac OFT consists of a simple tubular structure connecting the primitive ventricle to the aortic sac, lined by two major cushions: the parietal and septal outflow endocardial cushions. These OFT cushions are arranged in a spiralling pattern, reflecting the course of the aortic and pulmonary flows in the adult. These cushions, initially composed of extracellular matrix (ECM) called cardiac jelly, are populated by mesenchymal cells through endocardial-to-mesenchymal transition, regulated by Notch, bone morphogenetic protein, and TGF-β signalling. These OFT cushions also receive neural crest-derived cells in addition to endocardium-derived mesenchymal cells.6 The migration of neural crest cells into the heart is regulated by Rho-kinases, Wnt, bone morphogenetic protein, fibroblast growth factor, and semaphorin signalling.7 These cushions expand into the lumen and meet, initially fusing together. Subsequently, they merge with the aorticopulmonary septum, which is a protrusion formed by migratory cardiac neural crest cells from the dorsal wall of the aortic sac. Fusion of these components spirally grows into the OFT lumen, ultimately dividing the OFT into the aorta and pulmonary trunk in the 7th week of development (Figure 2A–2E).6
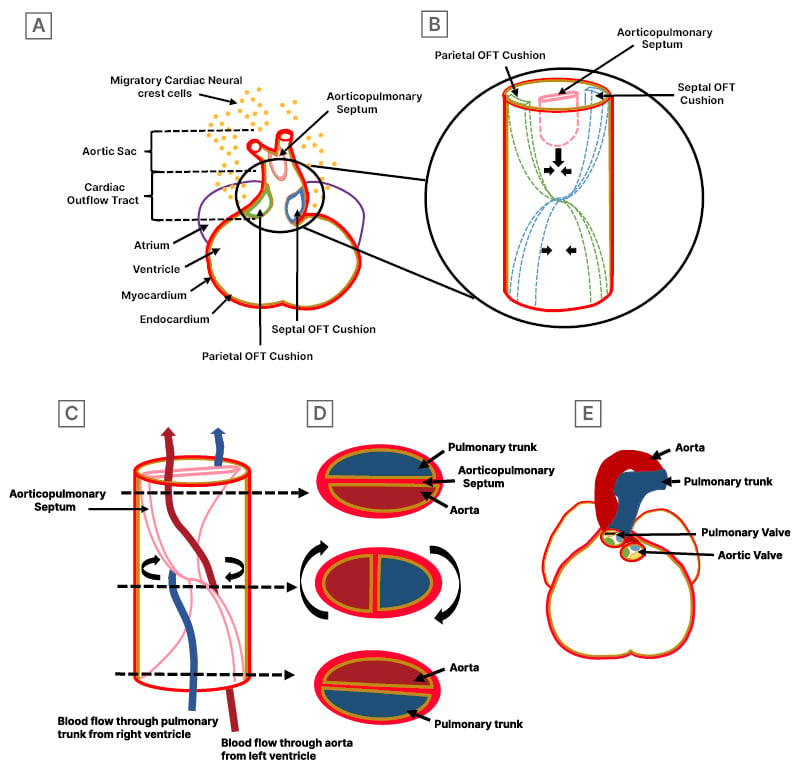
Figure 2: Embryology for an understanding of cardiac outflow tract septation development.
A) Formation of endocardial cushions and aorticopulmonary septum: the coronal section of the cardiac outflow tract shows endocardial cushion, known as parietal and septal OFT cushions, composed of extracellular matrix, which are populated by mesenchymal cells through the endocardial-to-mesenchymal transition and infiltration of cardiac neural crest-derived cell. Additionally, the section shows aorticopulmonary septum, originating from the dorsal wall of the aorta and composed of migratory cardiac neural crest cells. The zoomed-in circle view details the development of parietal and septal endocardial cushions and the aorticopulmonary septum. B) Formation of septation: OFT cushions lay in spiralling fashion and initially fuse together, subsequently merging with the aorticopulmonary septum. C) Spiral septation: the spiralling of aorticopulmonary septum results into the aorta and pulmonary trunk. The spiral aorticopulmonary septum separates the systemic arterial flow (red line) from the pulmonary arterial flow (blue line). The arrows indicate the levels of the sections shown in Figure 2D. D) Separation of the aorta and pulmonary trunk is complete. Sections through the newly formed aorta and pulmonary trunk, showing the position of aorticopulmonary septum and the relationship between pulmonary trunk and aorta, differ in the lower, middle, and upper tracts. E) The final position of the ascending aorta and pulmonary trunk in the 7th week of development, spiralling around as they depart from the heart, maintains their spiral relationship post-septation.
OFT: outflow tract.
In addition to the major cushion involved in OFT septation, two minor intercalated ridges also form in the atrioventricular junction and OFT, and they receive extracardiac contributions from cardiac neural crest cells. After OFT septation, semilunar valves develop from mesenchymal outgrowths from proliferation of the OFT cushions and intercalated ridges. These outgrowths undergo apoptosis to form cusps, then mature through matrix remodelling to create aortic and pulmonic valve primordia (Figure 3A–3C). These primordia subsequently develop by thinning, reshaping, and elongation, forming mature valve cusps with organised ECM layers of collagen rich fibrosa, glycosaminoglycan rich spongiosa, and elastin-filled ventricularis. Cardiac neural crest cells are found at the tip of the cusps, contributing to remodelling and maturation. Each type of endocardial cushions and intercalated ridges contribute to specific valve formations. Semilunar valve leaflets attached to the facing walls of the aorta and pulmonary trunk originate from major OFT cushions, while those attached to non-facing walls originate from left and right intercalated ridges. The parietal OFT cushion forms the right aortic and pulmonary valves, whereas septal OFT cushion forms the left aortic and pulmonary valves. Left and right intercalated ridge cushions form the anterior pulmonary and posterior aortic valves, respectively (Figure 4A–4C).6
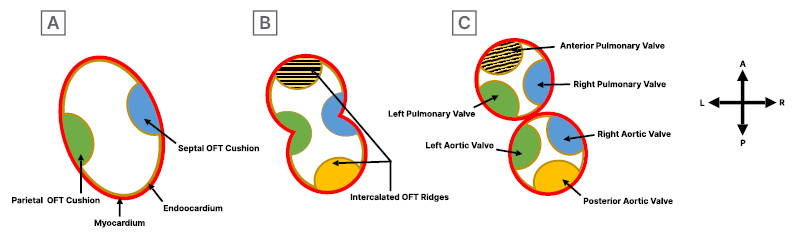
Figure 3: Development of semilunar valve in outflow tract of the heart.
A) The initial formation of the parietal and septal OFT cushions in the cardiac outflow tract. B) Formation of right and left intercalated ridges during early septation. C) Completion of septation and separation process, leading to the formation of semilunar valves. The parietal OFT cushion gives rise to the right aortic and pulmonary valve, the septal OFT cushion to the left aortic and pulmonary valve, and the left and right intercalated ridge cushions to the anterior pulmonary and posterior aortic valves, respectively.
OFT: outflow tract.
Genetic, transcriptional, and signalling disruptions can potentially lead to valve abnormalities, although the exact mechanisms of QPV formation remain unclear.6 Abnormal cusp formation appears to result from the atypical proliferation of mesenchymal cushions in the cardiac OFT and aberrant fusion of the aorticopulmonary septum.1 One study suggests that QPV may arise from the splitting of a valve cushion during early valvulogenesis in an animal model.8 Another study indicates that disrupted signalling Rho-associated protein kinase pathways in cardiac neural crest cells can lead to abnormal aggregation, potentially resulting in extra cushions and the formation of a quadricuspid valve in an animal model.9
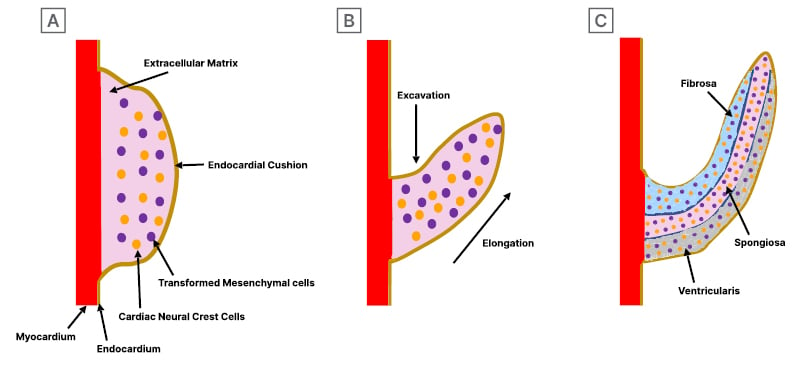
Figure 4: Development of semilunar valvular cusps.
A) Valve development begins with endocardial cushion formation, which becomes populated by mesenchymal cells through the endocardial-to-mesenchymal transition and infiltration of cardiac neural crest-derived cells. B) These cushions grow into primordial valves, undergoing excavation, thinning, and elongation, which leads to extracellular matrix (ECM) remodelling for reorganising ECM components in valve tissue. C) It concludes with the maturation of the semilunar valve, characterised by organised ECM layers of fibrosa, spongiosa, and ventricularis.
Although QPV often occurs as an isolated finding, such as in the case described above, some autopsy studies show an association of QPV with other congenital cardiac abnormalities such as patent ductus arteriosus, atrioventricular defects, atrial septal defects, and ventricular septal defects.5 Other case reports have described an association of QPV with significant pulmonary regurgitation, bicuspid aortic valve, stenosis and severe regurgitation, pulmonary artery aneurysm, and even sudden death.10-14 Although the authors’ case demonstrates an asymptomatic QPV, their understanding of how QPV contributes to cardiac pathology continues to evolve, and currently, its potential role in clinically detectable disease may be underestimated.
With the rise of radiology and its expanded application in assessing cardiac anatomy and function in adults, identification of QPV in living patients remains rare and often goes undetected due to limitations in TTE imaging (Table 1). The first possible explanation is that QPV is rarely associated with other cardiac anomalies, and is clinically quiescent. In such cases, there would be no clinical reason for obtaining cardiac imaging if QPV is the only defect during presentation without any symptoms.24 The second explanation is a technical challenge associated with TTE. Due to the anatomical disposition of the valve in relation to the thoracic wall, it is challenging to visualise its short axis accurately.16 The third explanation is that the innervation of the pulmonary valve remains relatively unaffected by ageing, while the innervation of the aortic valve is age-dependent and decreases in density with age.25 This may explain why the QPV is rarely linked to symptoms or significant abnormal valvular function. The fourth explanation is that, as in the case described above, the presence of four equal valve cusps results in equal stress distribution, complete valve coaptation with symmetrical overlap, and no flow disturbance. In contrast, patients with a small additional accessory cusp or varying size cusp could experience uneven stress distribution and abnormal cusp coaptation, potentially leading to progressive regurgitation. In one study, it was found that quadricuspid aortic valves with four equal sized leaflets are less prone to develop fibrous thickening and significant aortic regurgitation, while presence of an additional cusp smaller than the other, may lead to uneven stress distribution, fibrosis, and abnormal coaptation, potentially resulting in significant regurgitation.26
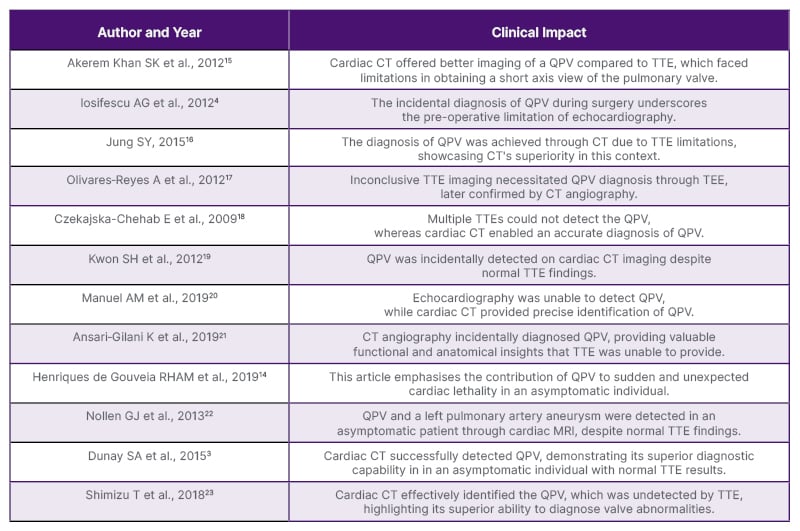
Table 1: A summary of past case reports and clinical impact on diagnosis of quadricuspid pulmonary valve.
QPV: quadricuspid pulmonary valve; TEE: transesophageal echocardiogram; TTE: transthoracic echocardiography.
Diagnosing QPV relies on imaging studies, with TEE, cardiac CT, and cardiac MRI being the most sensitive methods, offering enhanced visualisation of the pulmonary valve.3 These imaging techniques not only enable the observation of morphological characteristics of the valve, but also reveal associated structural deformities, whereas echocardiography mostly evaluates the functional abnormalities of the pulmonary valve. Cardiac CT and cardiac MRI are fast emerging as the most preferred imaging modalities for pulmonary valve diseases in adults, likely to result in QPV being identified in living persons much more frequently as these technologies are more universally adopted.27
QPV is often clinically silent, but it can be associated with congenital aortic valve anomalies due to shared morphogenesis of aortic and pulmonary valves, potentially requiring surgical intervention. The Ross procedure is widely acknowledged as effective in treating congenital aortic valve malformations by replacing the diseased aortic valve with patient’s own tricuspid pulmonary valve, followed by replacement of the pulmonary valve with a pulmonary homograft. This approach has been found to be effective in cases of congenital aortic valve malformations when a normal and functional tricuspid pulmonary valve is used. However, a QPV is not an ideal candidate for the Ross procedure due to its poorly understood haemodynamics.28 Despite promising early post-operative results, the development of second-degree autograft regurgitation within 4 years post-surgery suggests potential issues with using a quadricuspid pulmonary autograft in a Ross procedure. Therefore, pre-operative imaging assessment of pulmonary valve function and morphology is deemed necessary.29
Though QPV is underdiagnosed and rarely complicated, its identification is crucial, as it can avoid delayed diagnosis, which can lead to unrecognised cardiac complication based on variant type and cusp size, causing asymmetrical valve stress distribution, abnormal coaptation, and severe symptoms during stress conditions.4,23 Identifying anatomical variants can significantly impact clinical outcomes by reducing diagnostic delays. Lack of familiarity with anatomical variations may adversely affect medical, surgical, and imaging results, yet its importance in safe clinical practice is often overlooked.30 Enhanced awareness can facilitate early detection and effective management to prevent adverse clinical outcomes. This case report highlights a rare anatomical variant, offering valuable insights that contribute to medical knowledge, benefitting both education and clinical practice.
CONCLUSION
QPV is a rare congenital cardiac anomaly that is usually clinically quiescent and asymptomatic, and rarely presents with clinical complications. The authors reported on a rare case of QPV that was asymptomatic. QPV might seem unimportant, but its potential role in complications might be underestimated due to its benign nature, as it is mostly diagnosed post-mortem. Identifying anatomical variants can significantly impact clinical outcomes by reducing diagnostic delays. Failure to recognise variant anatomy may adversely affect medical, surgical, and imaging results. This case report aims to increase awareness and enhance clinical practice outcome of patients with anatomic variants.