
Abstract
Objective: This study compared sirolimus-eluting stents (SES) with everolimus-eluting stents (EES) in coronary artery disease patients.
Methods: A total of 1,174 patients were enrolled in the study; 290 patients (25.28%) were treated with EES and 884 patients (74.72%) were treated with SES. The trial (PRISM) was a randomised (in a 3:1 ratio), multicentre, single-blind, all-comers, single-arm, non-inferiority trial comparing SES and EES-implanted patients with coronary artery disease. The primary endpoint was a composite of safety parameters (including major adverse cardiac events [MACE], cardiac death, and myocardial infarction) and efficacy (parameters concerned to quantitative coronary angiogram). An intention-to-treat analysis was performed at 9 and 18-month follow-ups.
Results: The baseline characteristics were similar for both EES and SES groups. At the 9-month follow-up, MACE occurred in 5.86% and 2.43% of patients in the EES and SES groups, respectively. At the 18-month follow-up, this differential remained almost the same (i.e., 5.17 % of patients treated with the EES versus 2.14% treated with the SES). The rate of definite stent thrombosis at 9-month follow-up was lower in the SES group (11 patients [1.24%]) compared to the EES group (9 patients [3.10%]). At 18-month follow-up, the rate was 2.14% (19 patients) in the SES group and 4.13% (12 patients) in the EES group. When censoring the patients at the time of stent thrombosis, no significant differences between the two stent groups were found.
Conclusion: In this real-world trial, at 9 and 18-month follow-ups, SES (M’Sure-S) exhibited a better safety and efficacy profile when compared to EES in terms of MACE rates and definite stent thrombosis. However, the difference was not statistically significant and SES was found to be non-inferior to EES.
INTRODUCTION
Drug-eluting stents (DES) used for the treatment of coronary artery disease were an important development in the field of percutaneous coronary intervention (PCI). The use of balloon angioplasty and bare-metal stents resulted in an augmented rate of reocclusions and restenosis. The incidences of restenosis and target vessel revascularisations (TVR) are significantly reduced by DES compared to their antecessors, bare-metal stents.1,2 The second-generation DES, containing antiproliferative and immunosuppressive agents, are preferred over first-generation DES; this is due to the increased incidences of myocardial infarction (MI) and stent thrombosis observed following the use of first-generation DES.3 Despite second- generation DES being safer, both generations of DES offer equivalent levels of efficacy. These stents (second-generation stents) contain a cobalt chromium (L605) platform with ultrathin struts covered with a silicon carbide layer.4 In a study of sirolimus-eluting stents (SES), a first-generation DES exhibited demonstrable angiographic results versus an everolimus- eluting stents (EES).5 A study conducted by Han et al.6 examined the material characteristics and limitations of poly-L-lactic acid polymer- based bioresorbable scaffold in PCI. It also compared the strut thickness in bioresorbable scaffolds and metallic DES. The PRISM randomised control trial (RCT) aimed to determine the safety and efficacy of first-generation SES (M’Sure-S, Multimedics LLC, Ahmedabad, India) versus second-generation EES (EliminatorTM, Multimedics LLC).
The present study was designed to assess the procedural performance, angiographic result, and long-term clinical outcome obtained by M’Sure-S versus Eliminator. The safety and efficacy outcomes at the different time intervals of the PRISM RCT trial were compared, with a specific focus on long-term clinical performance of the study stents and cardiac events linked with definite stent thrombosis of SES and EES.
MATERIALS AND METHODS
Study Design, Recruitment, Enrollment, and Oversight
The PRISM RCT study was a prospective, randomised (3:1 ratio), open-label, all-comers, single-blind, non-inferiority, multicentre trial, with clinical follow-up at 9 and 18 months. The trial enrolled 1,174 patients; 884 patients were treated with SES and 290 patients were treated with EES. The multicentre study involved European and Indian populations. Patients >18 years old, presenting with symptomatic ischaemic heart disease and/or objective evidence of myocardial ischaemia, and ready for percutaneous transluminal coronary angioplasty, stenting, or emergency coronary artery bypass graft (CABG) were eligible for study participation. The mean age of the patients involved in the trial was 68 years. The research and development department of the PRISM RCT was responsible for data collection and monitoring. All source data were verified by independent monitors on site. All cardiac and non-cardiac adverse events were reviewed and monitored by a safety and data monitoring board. An independent clinical event committee adjudicated all clinical endpoints in a blinded fashion. The investigators involved in the study vouched for the accuracy of the data and analysis. Approval for the trial was granted by each of the institutional ethical committees at the participating sites. Informed consent forms were signed and collected from the participants prior to the study. The trial was registered in a clinical trials registry and conducted as per the ICH/E6/R1 guidelines. Unrestricted access to the data was given to the principal investigator post database lock and the decision was taken to prepare this manuscript for publication.
The most essential inclusion criterion was the presence of a de novo target lesion located in the native coronary artery suitable for conventional angioplasty and stenting, and that could be covered by one stent without overlapping. More precisely, the target lesion had to be present in the native epicardial coronary artery (2.5–4.0 mm in diameter) and had to be able to be covered by a single SES with a maximum length of 40 mm. Lesions with severe calcification, tortuosity, presence of thrombus, bifurcation sites, left main coronary artery involvement, saphenous vein grafts, and those with a left ventricular ejection fraction <30% were excluded from the study.
The main exclusion criteria were pregnancy; known hypersensitivity to or contraindication for sirolimus or any other mTOR; and hypersensitivity to or contraindication for aspirin, clopidogrel, or other thienopyridines. Additionally, hypersensitivity to or contraindication for cobalt, chromium, heparin, or contrast media that are routinely present during stenting procedures were exclusion criteria. Other exclusion criteria included pretreatment of target lesions by stenting methods, previous brachytherapy, presence of significant non-target lesions requiring treatment within 30 days of the index procedure, prior CABG to the target vessel, and acute MI within 48 hours.
Study Procedure
The study subjects were treated by routine angioplasty procedure as per previously published standard protocol with slight modification.4,7 Briefly, keeping the angiographic inclusion and exclusion criteria in mind, the stents were deployed upon receipt of visual estimation of the vessel diameter and lesion characteristics. At the end of the stent implantation, it was left to the interventional cardiologist’s discretion whether to treat the patient further with a post-dilatation balloon catheter. Dual antiplatelet therapy (aspirin concomitantly with clopidogrel) was continued for up to 1 year post-procedure. Procedural success was defined as a successful device implantation with a residual stenosis of <20% of the vessel diameter, event-free sheath removal, and subsequent discharge from the hospital. Angiographic follow-ups were performed at 9 and 18 months. Fractional flow reserve was used in cases of intermediate target vessel stenosis <70% with or without angina or >70% in the absence of angina during follow-up.
Quantitative Coronary Analysis and Clinical Follow-Up
All the coronary angiograms were analysed in an angiographic laboratory by automated software and independent technicians who were unaware of the clinical information and stent allocation pertaining to this study. The quantitative measurements included the in-stent lumen loss, in-segment late lumen loss, lesion length, percentage of diameter stenosis, and minimal luminal diameter (MLD). Follow-up was carried out at 9 and 18 months post-procedure. Follow-up information was collected either by a hospital visit or telephone contact with the patient or the referring physician. Patients were followed-up for up to a total of 18 months.
Study Endpoints
The primary safety endpoint was defined as major adverse cardiac events (MACE) at 30 days, defined as a composite of death, MI (both Q wave and non-Q wave MI), emergent CABG, or clinically driven target-lesion revascularisation (TLR) (repeat PCI or CABG). The primary efficacy endpoint was the in-stent and in-segment late loss at 9 and 18-month follow-up, respectively, determined by off-line quantitative coronary analysis (QCA) at the core laboratory. The secondary efficacy endpoints for the PRISM RCT were angiographic and device success, procedural success, QCA-derived vessel parameters in-stent, and 5 mm proximal and 5 mm distal from the edge of the stent (acute gain, MLD, diameter stenosis, late loss, binary restenosis, in-stent MLD pre, post, and at angiographic follow-up). Binary restenosis was defined as a diameter stenosis ≥50%.
Statistical Analysis
The trial was performed to assess the non-inferiority of SES to EES with respect to the primary endpoint (safety and efficacy) at 9 and 18 months. For superiority for all endpoints, 2-sided 95% confidence intervals (CI) and 2-sided p values were calculated. The primary analysis was performed according to the intention-to-treat principle. To compare the distributions of continuous variables between the study groups, the 2-sample student t test was used, and the chi-square method was used, with a power of 95% and significance level α=0.005, to measure any difference between the treatment arms. CI functions were computed considering death (and, in a sensitivity analysis, also stent thrombosis) as a competing risk. The MACE per patient was ranked according to the highest category on a scale ranging from 1) death, 2) MI, 3) CABG, to 4) TLR. Gray’s test was used for comparing the cumulative incidence functions and Cox regression to determine the cause-specific hazard ratios (HR). Patients treated with the SES were used as the reference group for overall and subgroup analyses for safety and efficacy parameters. HR was calculated for MACE at 18-month follow-up for prespecified patient subgroups (based on baseline demographic and clinical characteristics). A 2-sided p value <0.05 was considered statistically significant. Analyses were conducted using SPSS (SAS institute, Cary, North Carolina, USA).
RESULTS
Baseline and Procedural Characteristics
In this clinical trial, cumulative data comprised results from the 1,174 enrolled patients, 884 of whom were treated with a SES and 290 with an EES. The mean ages of the patients was ≤68 years, and among them male and female patients were bifurcated randomly, in a 3:1 ratio. The statistical analysis of other baseline characteristics like heart rate, previous myocardial infarction, stable angina acute coronary syndrome, diabetes, smoker, hyperlipidaemia, family history of coronary artery disease are represented in Table 1. Both groups achieved 100% of the procedural characteristics.
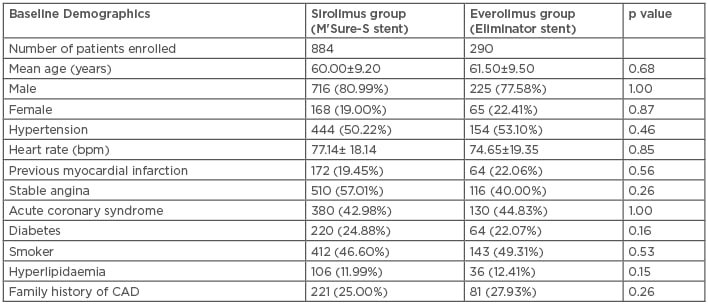
Table 1: Baseline characteristics of both sirolimus and everolimus-eluting stent treatment groups.
CAD: coronary artery disease.
Quantitative Coronary Analysis
QCA were obtained at four distinct timepoints: pre-stenting, post-stenting, at 9-month follow-up, and at 18-month follow-up. The pre-stenting QCA results showed lesion length (LL), median reference vessel diameter, minimum luminal diameter, and percentage diameter stenosis for SES patients were slightly higher when compared to EES (19.92 mm versus 18.41 mm, p value=0.93; 2.35 mm versus 2.20 mm, p value=0.10; 0.98 mm versus 0.97 mm, p value=0.82; and 79.29% versus 78.45%, p value=0.87, respectively). Here, LL for SES was shown to be non-inferior to EES.
Post-procedural analysis showed that in-stent residual diameter stenosis was slightly higher in the SES group (13.26%) compared to the EES group (12.78%). In-stent acute gain was measured to be 1.34 mm for SES compared to 1.35 mm for EES. In-segment analysis of the SES and EES groups revealed a MLD of 2.32 mm and 2.43 mm, respectively, and a residual diameter stenosis of 24.56% and 25.46%, respectively, with an acute gain of 1.34 mm and 1.43 mm, respectively.
At the 9-month timepoint, angiographic follow-up data from 874 and 282 patients were studied; median in-stent MLD for the SES and EES groups were found to be 2.30 mm and 2.20 mm, respectively, with a diameter stenosis of 11.26% and 11.21%, respectively, and a late lumen loss (LLL) of 0.05 mm and 0.04 mm, respectively. Binary restenosis was not found in any patients and this remained at 0.0% at the end of 9 months, thereby indicating the high efficacy of both the stents used in this trial.
There were no significant differences in angiographic measurements of lesions before and after the procedure. Angiographic follow-up at 18 months was performed in 1,119 patients (855 in the SES group and 264 in the EES group). The primary endpoint of the study, mean in-segment LLL, was 0.07 mm in the SES group and 0.10 mm in the EES group; thus, the results of the in-segment LLL met the criteria for non-inferiority of SES versus EES (non-inferiority margin=0.1 mm). The in-stent LLL showed similar findings; the mean in-stent LL was 0.06 mm and 0.08 mm for the SES and EES groups, respectively. Median in-stent MLD was found to be 2.17 mm, with a diameter stenosis of 14.26%; binary restenosis was not found in any patients and this remained at 0% after 18 months, thereby indicating high efficacy. Likewise, median in-segment MLD was 2.35 mm and thus the percentage diameter stenosis was calculated to be 21.24 mm, with no binary restenosis.
Major Adverse Cardiac Events and Stent Thrombosis
All patients in the trial achieved the primary endpoint and were followed for 9 months. At 9-month follow-up, MACE occurred in 22 patients (2.48%) who received SES and in 17 (5.86%) patients who received EES (HR: 0.98; 95% CI: 1.00–1.06; p=0.28). However, at 18-month follow-up, the occurrence of MACE was reduced to 19 (2.14%) and 15 (5.17%) in the SES and EES patients, respectively (Figure 1A).
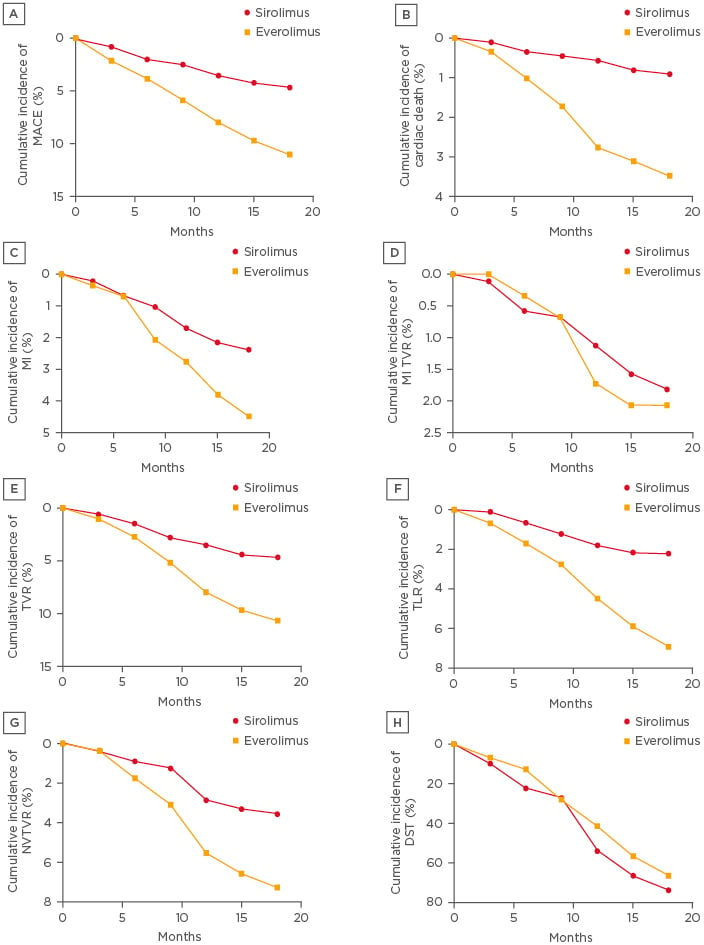
Figure 1: Cumulative event curves up to 18 months are shown for A) MACE, B) cardiac death; C) myocardial infarction; D) myocardial infarction target vessel revascularisation; E) target vessel revascularisation; F) non-target vessel revascularization; G) Non-target vessel revascularisation H) definite stent thrombosis.
DST: definite stent thrombosis; MACE: major adverse cardiac events; MI: myocardial infarction; NTVR: non-target vessel revascularisation; TLR: target-lesion revascularisation; TVR: target vessel revascularisation.
Initially, from 0–9-month follow-up, 4 patients (0.45%) and 5 patients (1.72%) died due to cardiac death in the SES and EES-treated groups (HR: 0.96; 95% CI: 0.97–1.07; p=0.47 [hl]Figure 1B[/hl]), respectively; furthermore, the same percentage of deaths was observed at 9–18-months follow-up in both groups. From the data it was observed that death rates in the SES group were lower compared to the EES group, however, the difference was non-significant.
MI was observed in 21 patients who received SES and 13 patients who received EES. At 9-month follow-up, 9 and 6 patients were found with MI who had received SES and EES, respectively (HR: 0.89; 95% CI: 0.99–1.02; p=0.25). The incidence of MI increased to 12 and 7 at 18-month follow-up, respectively (HR: 0.95; 95% CI: 0.99–1.03; p=0.29 [hl]Figure 1C[/hl]). However, SES-implanted patents had a lower incidence of MI compared to EES patients.
MI TVR was performed in 16 and 6 patients who received SES and EES, respectively (HR: 0.89; 95% CI: 0.98–1.03; p=0.36 [hl]Figure 1D[/hl]). At 9-month follow-up 6 and 2 patients were found to have MI TVR who had received SES and EES; but the incidence was raised to 10 and 4 at 18-month follow-up, respectively. There was a need for TVR in 47 and 31 patients in the SES and EES patient groups, respectively (HR: 0.92; 95% CI: 1.01–1.10; p=0.89 [hl]Figure 1E[/hl]). There was also a need of TLR in 20 patients with SES and in 18 patients with EES (HR: 0.98; 95% CI: 0.73–1.20; p=0.65 [hl]Figure 1F[/hl]). Non-TVR was also required in 31 patients with SES and in 21 patients with EES (HR: 0.97; 95% CI: 0.99–1.07; p=0.29 [hl]Figure 1G[/hl]). The total number of definite stent thromboses was lower in SES patients (30 [3.40%]) compared to EES patients (21 [7.23%]) (HR: 1.17; 95% CI: 0.99–1.02; p=0.24). The incidence of definite stent thrombosis (DST) did not differ significantly at 0–9-month follow-up (11 [1.24%] with SES versus 9 [3.10%] with EES; HR: 0.97; 95% CI: 0.99–1.02; p=0.10). However, at 18-month follow-up, DST was not significantly lower in the SES group than the EES group (19 [2.14%] versus 12 [4.13%] patients, respectively; HR: 1.03; 95% CI: 0.99–1.04; p=0.30 [hl]Figure 1H[/hl]). Results of the corresponding test for interaction were non-significant, which was deduced based on Cox regression analysis. Graphical representation of cumulative incidences of all the primary events of SES and EES implantation are shown in Figure 1.
DISCUSSION
Over recent decades, stent technology has gained momentum in PCI.8 The surface of the metal and the chemical properties of the materials play a pivotal role in designing an ideal, safe, and efficacious stent. In recent years, there has been a revolutionary change in stent material and design. The bare-metal stents prevent negative arterial remodelling in percutaneous transluminal coronary angioplasty, acute recoil, and closure of vessels.9 However, the use of bare-metal stents is limited by augmented instances of restenosis and repeat revascularisation and stent thrombosis rates.10-15 Thus, newer generation DES were developed that inhibit neointimal proliferations, reducing repeat revascularisation.
In this arbitrary, prospective comparison of SES and EES, the efficacy of SES in suppressing neointimal growth (expressed as LL) was non-inferior to the EES. Both stents showed exceptional LL profiles at 9 and 18-month angiographic follow-up. Clinical outcomes, including MI, cardiac death, and TLR, were typically similar between the two stent types, although this study was underpowered to demonstrate the variation in clinical outcome between the two stents. Moreover, the distinct and apparent DST rates were not statistically different between the two types of stents.
The outcome of this trial was demonstrating the non-inferiority of M’Sure-S versus Eliminator. The trial outcome of both EES and SES included low rates of DST and TLR. Previously developed DES are known to cause late stent thrombosis and have thick struts, which act as a barrier for early endothelialisation. SES has low strut thickness (59 μm), which promises early endothelialisation and reduces the risk of stent thrombosis. SES has drug-elution kinetics of 28–30 days and a polymer degradation that is short and well documented.16-19 SES was found to be safe and efficacious in preclinical models and in the primary safety and efficacy study.20
In terms of efficacy parameters, at 9-month angiographic follow-up, 0.05 mm of in-stent medial LL were observed and no binary restenosis was recorded. Analysis of data demonstrated safety and efficacy of SES in line with other published randomised trials.21
This study demonstrated a high safety profile of DES, with 13%, 10%, and 0% of patients exhibiting MACE, stent thrombosis, and binary restenosis, respectively. Data from other similar studies demonstrated that, in comparison to EES, SES had a better enduring safety and efficacy report.10,22,23 These results also revealed a 14% reduction in MACE rate in SES patients, which was largely due to a lower risk of very late DST. During 0–9 months, MACE rate did not differ significantly, which is the main prevalent timepoint considered for determining the primary endpoint in head-to-head contrasts of DES. However, from 9–18 months the MACE rate was non-inferior with SES versus EES.
Throughout the 18 months, the liability of DST was sporadic and alike in both groups. In a comparison of first-generation DES (SES and EES), the initial pre-eminence of the EES was lost at 18-month follow-up. DST occurred with an annual rate of 1.24–3.10% for both stent types.24-26 In this trial (SES versus EES), DST was more recurrent after implantation of EES than after SES, although contradictory results were recorded between 9 and 18-month follow-up. The present results from the PRISM RCT with follow-up to 18 months indicated that the composite endpoint safety and efficacy factors of SES versus EES were not significant. The rates of DST also presented similar results, with both being equally efficacious, as per studies.27 In this PRISM RCT trial, SES patients exhibited better safety and efficacy profiles in MACE rates and DST when compared to EES patients. However, the difference found was not statistically significant. Like most stent trials, the PRISM RCT trial was designed as a single-blinded study, and the authors believe that the scarcity of double-blindness would not influence the results, as all endpoints were objective and determined by an event committee that was blinded to treatment group assignment during the adjudication process. This registry-based randomised clinical trial design has been used in all PRISM RCT trials and has established significant attention as a way of experimenting comprehensive, self-regulating clinical trials. Advantages of this approach include a substantial reduction in the expense associated with a randomised trial because an established registry infrastructure was able to be used. Moreover, the study design provides data that are more comparable to real-life situations because of the absence of study-related interventions.
CONCLUSION
At 9 and 18-month follow-up, SES established a better safety and efficacy profiles, and the difference in MACE rate between SES and EES-treated patients was non-significant. This effect was largely due to a lower risk of DST.





