Abstract
In Part One of this exploration of the pathogenesis of coronavirus disease (COVID-19), the author will evaluate the viral and cellular immunological basis for the condition. The virus demonstrates a remarkable capability not just to evade, but to exploit host immune characteristics to perpetuate viral replication. In this regard, severe acute respiratory syndrome (SARS)/severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) disables most antiviral mechanisms, including the early interferon response, and avoids detection to permit unimpeded viral multiplication. Consequently, antigen-presenting cells fail to adequately stimulate the T-cell receptor. As a consequence, T-cell p53 remains highly expressed, which in turn disables an adequate effector T-cell response.
Replicating SARS-CoV-2 double-strand RNA robustly activates protein kinase R (PKR)/PKR-like endoplasmic reticulum kinase (PERK). While the virus is grossly invulnerable to its antiviral effects, PKR is crucial for effecting the cytokine milieu in COVID-19. PERK is a component of the unfolded protein response, which eventuates in autophagy. SARS virions use double-membrane vesicles and adapt PERK signalling not only to avoid autophagy, but to facilitate replication. Viral activation of PKR/PERK is mutually exclusive to NLRP3 stimulation. The NLRP3 pathway elaborates IL-1β. This is chiefly a feature of paediatric SARS/SARS-CoV-2 cases. The difficulties encountered in predicting outcome and forging effective therapeutics speaks to the breadth of complexity of the immunopathogenesis of this virus.
INTRODUCTION
Severe and fatal severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection is characterised by chiefly pulmonary manifestations. Clinically, pneumonias have been subdivided into specific phenotypes: a spectrum from patchy ground-glass opacification to the oedematous lung with atypical acute respiratory distress syndrome features.1Bilateral diffuse alveolar damage with cellular fibromyxoid exudates and desquamation of pneumocytes with hyaline membrane formation are pathologically apparent.2 For the majority of patients (varying by age and other factors), the condition has been mild. It appears that the causative virus, SARS-CoV-2, evolved into two major genomic types (L and S types), with a roughly 70:30 split.3 Although the L-type is likely phenotypically more aggressive, variability in host response is clearly a major determinant of outcome. Studies of SARS-CoV-2 have been limited by the novel nature of the virus. Nonetheless, valuable insight may be drawn from existing studies on the biology and pathogenesis of the SARS virus because of the significant sequence homology.4
VIRUS-SPECIFIC CHARACTERISTICS: ESTABLISHING INFECTION
Extrapolating from SARS-CoV virology, initial investigations implied that SARS-CoV-2 used angiotensin-converting enzyme 2 (ACE2) for cellular entry.5 ACE2 expression is predominantly, but not limited to, pulmonary tissue (Type 2 pneumocytes and ciliated airway epithelial cells), with expression of SARS virus previously reported in extrapulmonary ACE2+ tissues.6-9 SARS spike proteins (S-proteins) were found to bind robustly to ACE2.10,11 Recent reports suggest that the nature of SARS-CoV-2 affinity for ACE2 is 10-times more avid than that of SARS. TMPRSS2, a serine protease, is used for S-protein priming within cells, which is essential for viral spread and (especially pulmonary) pathogenesis in the infected host (cathepsins B and L may also be used but are inessential for this purpose).5,12 A further portal of entry is cell surface expression of cluster of differentiation 147 (CD147), also known as basigin or extracellular matrix metalloproteinase inducer (EMMPRIN).13-15
Of the large coronaviral genome, the open reading frames 1a and 1b (ORF1a and 1b), encoding the viral replicase, comprise two-thirds. The replicase includes multispanning transmembrane proteins that physically anchor the replication/transcription complex to the intracellular membranes. Within the cytoplasm, double-membrane vesicles (DMV) branch off from the rough endoplasmic reticulum (ER), reminiscent of autophagosomes.16 The coronavirus endonuclease, which resides in the replication complex, prevents simultaneous activation of double-strand RNA (dsRNA) sensors melanoma differentiation-associated protein 5, 2’-5’-oligoadenylate synthetase 3, and protein kinase R (PKR). This strategy permits evasion of the host innate antiviral defenses.17 Thus, viral kinetics are rapid in the early period (first 48 hours) post-infection.
THE IMMUNOLOGICAL RESPONSE IN SEVERE COVID-19 INFECTION
Severe SARS/SARS-CoV-2 is suggestive of an apparent biphasic (dysregulated) immune response.18 A weak or absent interferon Type 1 ([IFN-1] i.e., IFNα and IFNβ) response during the early phase of SARS-CoV-2 infection plays an important role in permitting viral replication within nasopharyngeal cells and pneumocytes.19,20 It is apparent that virion nonstructural protein 1 within infected cells can suppress host gene expression (including IFN-1), promote host messenger RNA (mRNA) degradation, and inhibit host protein translation.21 Furthermore, the SARS coronavirus papain-like protease induces inhibition of the production of IFN-1 and proinflammatory cytokines in toll-like receptor 3 (TLR3), retinoic acid-inducible gene 1, and TLR7 signalling pathways, thus disabling detection mechanisms in the endosome and in the cytoplasm.22-26
Indeed, some investigators could find no or only modest evidence of IFN response to SARS infection.27,28 However, fatal/severe SARS in humans was accompanied by a late but robust and persistent expression of IFN-1, especially from plasmacytoid dendritic cells. Concurrent expression of IFN-1 and IFN-stimulated genes appear to preclude adequate T-cell and antibody responses;18,29-31 this was demonstrated in SARS-infected IFNαβ receptor knockout (IFNAR-/-) mice, who exhibited only mild to moderate weight loss and clinical disease. These mice demonstrated minimal alveolar oedema and increased peribronchiolar/perivascular immune cell infiltration, which resolved with viral clearance by 10 days post-infection. By contrast, BALB/c mice with intact IFNAR exhibited a six-fold increase in (mainly) inflammatory monocyte-macrophages by Day 3 post-infection, which is too late for the peak of viral replication. This was then abrogated in the absence of IFN-1.32
Augmented T-cell apoptosis in SARS infection impedes T-cell response and engenders a relative lymphopaenia.33Normally, IL-2 promotes the differentiation of effector T cells in the presence of antigen-specific T-cell receptor (TCR) stimulation. TCR engagement by peptide-bound major histocompatability complex (MHC) molecules on antigen-presenting cells (APC) changes the topology of the TCR, inducing the formation of microclusters at the T cell–APC interface, named the immunological synapse. This arrangement helps to improve T-cell antigen recognition.34
However, in SARS, antigen-specific TCR expression is weakened by an early hyporesponsiveness of APC, especially in dendritic cells. SARS-CoV does not upregulate the expression of CD83, CD86, MHC Class I, or MHC Class II molecules on immature dendritic cells.35 Also, the late surge in IFNβ alters histone modifications in the IL-2 promoter to retain the locus in an inaccessible configuration, thereby curbing the T-cell response.36 Any elevation in IL-2 expression, secondary to immune monocyte-macrophage stimulation of naïve T cells, induces a sustained increase in p53 protein expression in the T cell. Downmodulation of p53 is necessary for antigen-specific responses of naïve and antigen-primed peripheral T cells and T-cell clones. This prevents proliferative TCR signalling, critical for antigen-specific CD4(+) T-cell responses, despite adequate signalling through the IL-2 receptor.37 It is this failure to recruit CD4+ T cells to the lungs (and not CD8+ T cells) that adversely affects viral clearance. The absence of the CD4+ T-cell response leads to enhanced interstitial pneumonitis.18 Furthermore, respiratory tract memory CD4+ T cells, in conjunction with IFNγ, would provide a more robust intermediate-term defence against reinfection.38 The latter may account for the inconsistent nature of postinfective antibody production.
T cells can attenuate cytokine storms by suppressing the immune response.39 It is the impairment of the regulatory mechanisms of T cells in a cytokine-rich milieu that fosters the development of the cytokine storm.40 In view of the abortive nature of T-cell infection by SARS-CoV-2, it is unlikely that this, per se, makes a significant contribution to the observed lymphopenia in coronavirus disease (COVID-19) infections.41
DISEASE-SPECIFIC IMMUNOLOGY
The Role of Protein Kinase R
While the impairment of host gene expression undoubtedly plays a part in infected epithelial cell apoptosis, much of the apoptosis appears to be secondary to host PKR expression, which is engaged as a stratagem to detect replicating dsRNA. PKR induces phosphorylation of elongation initiation factor 2α (eIF2α) which, in turn, blocks protein synthesis through translation of mRNA. The phosphorylated eIF2αalso encodes for antiviral factors and mediates the integrated stress response.41 The blockade of protein synthesis results in the decrease or prevention of viral replication, and may result in apoptosis.42 PKR can also induce apoptosis independently of eIF2α phosphorylation, by activation of the FAS-associated death domain (FADD)/caspase-8/caspase-3, and caspase-9 apoptotic protease-activating factor-1 (APAF-1) pathways.43-46 In spite of a significant burden of PKR expression, SARS-CoV is not susceptible to its antiviral activities. Indeed, rather than inhibiting PKR activation, translation of SARS-CoV mRNA proceeds despite eIF2α phosphorylation.42
PKR increases IFN1 expression via an eIF2α-independent mechanism by regulating IFNα/β mRNA stability.41,47 As well as contributing to the development of interstitial pneumonia, this spike in IFN1 creates an autoamplifying loop, given that IFN1 is known to enhance PKR expression.42 Activation of PKR by dsRNA has been shown in several cell types, including airway epithelial cells, to result in phosphorylation of IκB and therefore activation of NF-κB.48-51 p38 mitogen-activated protein kinase (MAPK) expression requires PKR.52,53 dsRNA induction of TNF-α requires PKR activation, but IL-1β induction follows a PKR-independent pathway.54 IL-6, IL-8, and Regulated upon Activation, normal T-cell expressed and presumably secreted (RANTES), among others, are promoted by PKR expression.42 Exuberant induction of PKR is the substantial driving force behind the cytokine-rich milieu observed in SARS/SARS-CoV-2 infection (Figure 1).
Note should be made here that levels of PKR may be already elevated chronically in individuals by virtue of a string of host factors including obesity, ageing, diabetes, congestive heart failure, cancer, and genetic factors (e.g., ApoE4, the most prevalent risk factor for Alzeihmer’s disease), all of which have been found to yield worse outcomes in SARS-CoV-2 infections.55,56
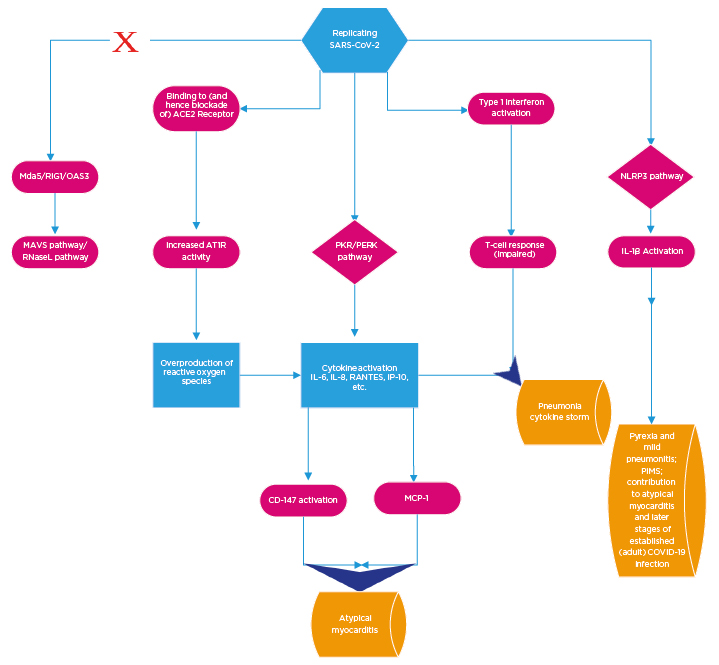
Figure 1: Overview of the basic cellular mechanisms contributing to coronavirus disease (COVID-19) pathogenesis.
Viral S protein binding to ACE2 receptors results in preferential activation of AT1R. The net consequence of this is a local overexpression of reactive oxygen species. In adults, the PKR/PERK pathways are preferentially activated to the exclusion of other antiviral pathways. This leads to the upsurge incytokine production. Subsequent consequences of this include activation of CD147 and MCP1, which contribute to atypical myocarditis. SARS CoV-2 initially disables the Type 1 interferon response but, subsequent to peak viraemia, Type 1 interferons are produced to excess. This impairs T cell response. The pro oxidant milieu in conjunction with the cytokine excess and blunted T cell response culminate in COVID-19 pneumonia and cytokine storm (across a spectrum of clinical severity). The NLRP3 pathway appears to be active early in paediatric infections to the exclusion of PKR/PERK (and at a later stage in adult disease). The clinical consequences include a relatively milder pneumonitis and pyrexia and rarely, in extreme circumstances, PIMS. It also contributes to the later stages of infection in adults.
ACE2: angiotensin converting enzyme 2; AT1R: angiotensin 1 receptor; RIG1: retinoic acid inducible gene I; MAVS: mitochondrial antiviral signaling protein; MCP1: monocyte chemoattractant protein 1; Mda5: melanoma differentiation associated protein 5; OAS3: oligoadenylate synthetase; PERK: protein kinase R endoplasmic reticulum kinase; PIMS: paediatric multisystem inflammatory syndrome; PKR: protein kinase R; RANTES; regulated upon activation, normal T cell expressed and presumably secreted; RNaseL: ribonuclease L; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
The Role of NLRP3 Inflammasome (Paediatrics)
Paediatric SARS patients were demonstrated to have markedly elevated circulating IL-1β levels. This suggests selective activation of a caspase-1-dependent pathway. Notably, only mild/minimal activation of IL-6 and TNF-α was noted in paediatric populations.57 However, as indicated above, IL-1β is independent of the PKR pathway. Rather, IL-1β is a caspase cleavage product of the nucleotide-binding domain and leucine-rich repeat and NLRP3 pathway.58 It appears that the SARS-CoV ORF8b robustly activates the NLRP3 inflammasome by providing a potent signal 2, required for activation. Note that inflammasome activity requires signals to effect cleavage: signal 1 is a priming signal and signal 2 is an activation signal. Mechanistically, ORF8b interacts directly with the leucine-rich repeat domain of NLRP3 and localises with NLRP3 and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) in cytosolic dot-like structures. ORF8b triggers cell death consistent with pyroptotic cell death in macrophages; while in those cells lacking NLRP3, accumulating ORF8b cytosolic aggregates cause ER stress, mitochondrial dysfunction, and caspase-independent cell death.59 Likewise, SARS-CoV ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC.60
It has been shown that PKR can suppress the NLRP3 inflammasome activation by modulating the microtubular cytoskeleton.61,62 It is evident, therefore, that a key mediator of the differential clinical presentation apparent between the paediatric population (who almost universally experience a mild or at worst moderate infection) and older adults (especially those with the aforementioned comorbidities, who are more predisposed to severe illness with COVID-19) is whether or not the NLRP3 pathway is activated in preference to a mutually exclusive pathway involving PKR overexpression.
The relatively rare occurrence of the Kawasaki-like syndrome as part of the spectrum of paediatric presentation with COVID-19, known as paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 infection in Europe or multisystem inflammatory syndrome in children in the USA, bears the hallmarks of an autoinflammatory syndrome/vasculitis, whose pathogenesis owes to IL-1β overexpression.63,64
The Role of Protein Kinase R Endoplasmic Reticulum Kinase
PERK is also elaborated as a consequence of dsRNA presence within the cytoplasm. PERK is a sensor in the unfolded protein response pathway.65 PERK is further activated by the SARS-CoV 3a protein. As well as causing eIF2α phosphorylation, it stimulates expression of ER molecular chaperones such as glucose-regulated protein 78 (GRP78) and GRP94.66 Increased transcriptional activation and biosynthesis of ER chaperones would enhance folding of the 3a and other viral proteins in the ER lumen. ER-associated degradation (ERAD) will normally delete an increased load of unfolded and misfolded proteins within the ER.67 This requires activation of the Inositol-requiring enzyme-1/X-box binding protein-1 and activating transcription factor 6 pathways. However, by activating PERK only, and not these ERAD components, the SARS-CoV 3a protein is able to enhance folding of common viral structural proteins S, E, M, and N, while avoiding activation of ERAD (which would be detrimental to virion assembly and likely trigger autophagy-dependent cell death).68
SARS-CoV must traffic across the ER membrane, thereby forming structures called double-membrane vesicles (DMV), which are thought to provide the necessary platform for the viral replication process while avoiding immune detection.69DMV, which originate from the ER membrane, contain nonstructural transmembrane proteins (nsp)3 and nsp4 and viral dsRNA,70,71 but lack markers typical for the ER Golgi intermediate compartment or Golgi.72
DMV are coated with microtubule-associated protein light chain 3 (LC3), which is a ubiquitin-like modifier.73 These ubiquitin-like modifiers recognise specific receptors that target associated vesicles to particular cellular locations.71,72LC3 can exist in in a lipidated (LC3-II) or a nonlipidated form (LC3-I) form. LC3-II is involved in fusion of autophagosomes to lysosomes/proteomes,74 but coronavirus DMV display the nonlipidated LC3-I and thereby evade destruction.71
In overview, it is clear that the PKR/PERK pathway is used for propagation of the viral lifecycle and is central to the cytokine-driven pathogenesis of the disease. This acknowledgement is not an assertion that PKR/PERK is the only pathway active in severe COVID-19 in adult patients. SARS-CoV-2 is a positive-sense single-stranded RNA virus that activates PKR/PERK during the replicative process when dsRNA is present. When viral replication is reduced, the NLRP3 inflammasome can become active, particularly in later stages of infection. In addition, the NLRP3 caspase cleavage product, IL-1β, may be elaborated via noninflammasome means (e.g., during a protracted inflammatory process via release of neutrophil serine proteases).
THE CONTRIBUTION OF CD147 TO SEVERE DISEASE PATHOGENESIS
Further to the exploitation of the PKR/PERK pathway, SARS-CoV-2 may also use the inflammatory milieu as a means to further its own cell invasive/replicative potential. For instance, IL-6 has previously been demonstrated to promote expression of CD147, a key receptor for viral cellular entry.75-77 This may be a normally adaptive process in the host since its expression may, in conjunction with cyclophilins, contribute to the recruitment of immune cells to sites of inflammation via chemokine‐like activity.78 Cyclophilins have previously been found to contribute to coronavirus pathology. Indeed, overexpression of SARS-CoV nonstructural protein 1, as well as infection with live SARS-CoV, strongly increased signalling through the calcineurin/nuclear factor of activated T cells (NFAT) pathway and enhanced the induction of IL-2. The latter is compatible with the immunopathogenicity and cytokine dysregulation observed in severe SARS cases.79
CD147/EMMPRIN is a widely expressed integral plasma membrane glycoprotein that is expressed at varying levels in many cell types, including haematopoietic, epithelial, and endothelial cells.80-82 Its cell surface expression (with cyclophilin cofactors) has been associated with interactions with extracellular matrix proteins, such as matrix metalloproteins, and integrins.83,84 While it is appreciated that IL-6 alone can be responsible for induction of thromboinflammation, it appears that CD147 overexpression is also proficient in this role, eliciting leukocyte chemotaxis and adhesion, as well as platelet activation and subsequent thrombus formation through the binding of various interaction partners.85-87 CD147 may assist IL-6 with the intravascular expression of vitronectin, plasminogen activator inhibitor-1, and von Willebrand factor, which forms the prothrombotic phenotype in severe SARS cases.88,89
THE VIRAL INDUCTION OF OXIDATIVE DAMAGE
The SARS-CoV-2 spike protein binds with great avidity to the ACE2 receptor.12 Once the protein has been cleaved by the serine protease TMPRSS2, the virus can be endocytosed, likely via micropinocytosis.90 Another serine protease, ADAM-17, is activated upon binding of SARS-CoV to ACE2 and facilitates viral entry. In fact, knockdown of ADAM-17 by small interfering RNA severely attenuates SARS-CoV cellular entry. ADAM-17 functions as a TNF-α cleavage enzyme and thus it contributes to the presence of TNF-α in SARS pneumonia.91 Pulmonary endothelial expression of ACE2 is lower in older adults relative to younger individuals.92 This would appear to imply that older adults should be less susceptible to contracting the virus (at least at a cellular, biological level) and has also served as something of a paradox in that patients with chronic obstructive pulmonary disease who actively smoke have higher levels of ACE2, but have had relatively low mortality compared to those with cardiovascular disease who tend to have relatively low expression of ACE2.93,94Angiotensin II levels have been demonstrably high in patients with COVID-19, thus apparently vindicating the theory that viral sequestration of ACE2 has a biochemical consequence for the host.95 In spite of this, patients who contract COVID-19 are not rendered overtly hypertensive. Moreover, the renin–angiotensin system is activated in almost any physiological stress scenario.96
In the absence of ACE2, signalling via the angiotensin I receptor is enhanced, apparently contributing to lung injury and pulmonary oedema in SARS.97,98 Angiotensin II binding to the angiotensin I receptor mediates its adverse effect on the lung through various subtypes of NADPH oxidase to produce reactive oxygen species.99 Furthermore, NADPH oxidase enhances phosphorylation, and hence activation, of PKR.100 The specific pathogenic contribution of ACE2 sequestration to COVID-19 pneumonia has been reviewed elsewhere.101
Haem oxygenase-1 (HO-1) is a stress-inducible, anti-inflammatory, and cytoprotective enzyme expressed in most cell types in organisms. Under several stress stimuli, HO-1 expression and activity is upregulated to catalyse the rate-limiting enzymatic step of haem degradation into carbon monoxide, free iron, and biliverdin.102 Besides its effects on cell metabolism, HO-1 is also capable of modulating host innate and adaptive immune response to sepsis, transplantation, and autoimmunity, and prevents oxidative damage associated with inflammation. HO-1 can exert a significant antiviral activity against a wide variety of viruses.103 Its activation decreases the migration of polymorphonuclear leukocytes to the lung. This may, in certain circumstances, reduce oxidative tissue damage.104 Crucially, it has been demonstrated to inhibit dendritic cell activation and immunogenicity.105 Failure to recruit functional respiratory dendritic cells to the lungs has been identified as a key defect, permitting the pathogenesis of SARS-CoV-induced pneumonia.106,107 This may be pertinent in the early stages of SARS/SARS-CoV-2 pneumonia.
In myeloid cells, HO-1 forms a complex with interferon regulatory transcription factor 3 (IRF3), which is required for IRF3 target genes and alters responses in infected cells.106 In fact, HO-1-deficient macrophages will show reduced expression of IFN-β and IRF3. SARS coronavirus papain-like protease inhibits IRF3 activation at a step after phosphorylation, which is dependent on the deubiquitination activity of papain-like protease.109 In later stages of the infection, caveolin-1 overexpression in lung epithelial cells will competitively inhibit HO-1 (caveolin-1 will be discussed further in Part Two).110
FUTURE DIRECTIONS: USING PATHOGENESIS TO BUILD DRUG PLATFORMS
The PKR/PERK pathway is the pathogenic motive force behind the severity of COVID-19. The NLRP3 inflammasome fuels an alternative, mutually exclusive, and apparently more benign condition. It follows that blockade of the PKR/PERK pathway would be distinctly advantageous to the host. While the expectation would be that it would not prevent infection, the nature of the infection would be much less severe.
To date, several pharmacological inhibitors of PKR have been investigated with varying degrees of adequacy. It is clear that identification of an inhibitor of PKR with good drug properties is an urgent necessity. There are some useful articles assessing this specific topic.111-114 N-acetylcysteine has been demonstrated to partially remediate the apoptotic consequence of PKR activation and oxidative stress.100 Furthermore, N-acetylcysteine has been demonstrated to alleviate the cytokine overproduction that occurs in alveolar Type II cells in the context of other respiratory viral infections such as influenza A and B and respiratory syncytial virus.115 N-acetylcysteine accomplishes this through inhibition of NF-κB translocation to the nucleus and phosphorylation of p38 MAPK, both of which are part of a stress pathway induced by PKR to promote IL-6, IL-8, and monocyte chemoattractant protein 1 overproduction.115-118
Pharmacological inhibitors of CD147 are also being developed for heterogenous indications.119-120 There may also be some rationale behind use of the humanised anti-CD147 IgG2 monoclonal antibody, meplazumab, which has been licensed as an orphan drug by the U.S. Food and Drug Administration (FDA) for treatment of malaria to counter not just viral entry into cells, but also the deleterious effects induced by CD147 in severe COVID-19.121 However, pharmacological evaluation is beyond the scope of this paper.
CONCLUSION
In conclusion, the author offers a rational breakdown of the cellular host–viral interaction in COVID-19. A substantial component of this analysis is extrapolated from the laboratory work already done in the field of SARS-CoV and other coronaviral infection/pneumonia. In Part Two, the author will probe the key mediators of specific systemic pathogenicity in COVID-19. It is the author’s strong recommendation that the details presented herewith are made subject to scrutiny in detailed animal studies involving SARS-CoV-2. Furthermore, the investigation of the platforms for drug treatment require accelerated human trials to establish efficacy and to curb the ongoing toll of lives lost and economic damage caused by this pandemic.