Abstract
Epitope identification is a key step in vaccine development, and this can be achieved much faster and less expensively with in silico methods, compared to traditional methods for vaccine production. In silico methods applied in this research utilised both bioinformatics and immunoinformatics approaches for chikungunya virus vaccine design, which involved the retrieval of sequences from databases, and identification of conserved regions within the sequences by multiple sequence alignment on the MEGA X software (Pennsylvania State University, State College, USA). The epitopes in the conserved regions were selected, and various immunological predictions and screenings were carried out by employing immunological databases and tools. This process identifies epitopes such as conservation of cytotoxic T lymphocyte, helper T lymphocytes, and B cell epitopes. The primary, secondary, and tertiary structure of the vaccine was also predicted using structure predicting servers, and finally, the vaccine candidate was docked to toll-like receptor 4 to study its binding affinity and configuration.
A total of 125 conserved antigenic epitopes were selected from capsid, 6K, and E1 proteins, which were found to be non-allergens and conform to acceptable physicochemical standards, as reported by other authors with similar work. The epitopes were predicted to be capable of inducing cytotoxic T lymphocytes, helper T lymphocytes, and B cell production. Construction of secondary structure was done using the Self-Optimized Prediction Method with Alignment (SOPMA), which predicted 17.96% α-helices, and 4.69% β-turns, among others. Predicting the tertiary structure provided five models, of which Model 1 was selected on the bases of its confidential score of 0.59, estimated TM-score of 0.79±0.09, and root mean square deviation of 8.0±4.4Å. Validity analysis revealed a Ramachandran plot where 97.2% of the vaccine residue was within the favoured region, and the peptide showed a Z-score of -1.52. The predicted peptide effectively docked with toll-like receptor 4 with a binding energy of -1,072.8. From the data obtained, it was revealed that the selected epitopes are highly immunogenic, non-allergenic, conform to native protein, and form a peptide capable of vaccine application. The authors can conclude this is a promising candidate for vaccine design and development.
Key Points
1. Chikungunya fever, first reported in East Africa, now affects over 110 countries globally. Without a cure, emphasis on prevention is crucial to combat the spread of this debilitating and neglected tropical disease.
2. The research focuses on virtual identification of immune-stimulating epitopes within the chikungunya virus protein, which have promising properties for the design and development of vaccines against infection.
3. Preventive measures are crucial in controlling infections such as chikungunya fever, given its gradual progression towards pandemic status. Identifying and implementing preventive strategies is key to effective management.
INTRODUCTION
Chikungunya fever (CHIKF) is a viral disease that is similar to dengue fever. It is a key public health concern in tropical and sub-tropical countries such as Kenya, where it re-emerged in 2004 and caused one of the largest epidemics on record.1 The causative pathogen is the chikungunya virus (CHIKV), belonging to the genus Alphavirus of the family Togaviridae. It is an enveloped virus transmitted between humans by two species of mosquitoes (Aedes aegypti and Aedes albopictus).2 CHIKV is comprised of a positive-strand RNA virus genome that is approximately 11.8 kb long with two open reading frames (ORF). The first ORF codes for four non-structural peptides responsible for genomic RNA replication and RNA transcription to structural proteins; and the second ORF codes for the structural protein C-E3-E2-6K-E1, which cleaves to form structural proteins such as capsid (Cp) and E3-E2/TF-E1.3
CHIKV replicates within the fibroblast or macrophages at the site of inoculation after it has been deposited into the skin or blood of a host by an infected mosquito’s bite. The virus spreads from the inoculation site through the lymphatic system and blood stream to other sites of replication, such as the lymphoid organs, muscles, peripheral joints, and tendons, within 2–4 incubation days. After this, a sudden manifestation of clinical symptoms that include headaches, rigors, incapacitating joint pain, high fever, photophobia, and petechial rash occurs. CHIKV has been reported to reach the brain and liver in very severe cases, leading to a more life-threatening condition.4
Acute phase symptoms of CHIKV infection can typically last from a few days to weeks, while the duration of symptoms for chronic phase disease has not been extensively studied. Report of recurring joint pain lasting for years has been recorded among 30–40% of infected patients; but this is suggested to be mediated by immune response and not as a result of chronic infection.5
Currently, there are no drugs for treating CHIKV infection, despite the fact it was declared an epidemic in over 14 countries by the World Health Organization (WHO) by the end of April 2014.6 In the absence of any known therapeutic, preventive measures such as a vaccine should be considered the best alternative to controlling the spread of the disease, as the virus is limited in its variety of antigen, and lifelong immunity is impacted for individuals who have suffered infection.7
The search for a CHIKF vaccine started just after the first outbreak in the 1960s, and this has resulted in the development of various types of vaccines, including live-attenuated vaccines, inactivated vaccines, chimeric vaccines, recombinant virus-vectored vaccines, and subunit vaccines.8
Subunit vaccines are vaccines that primarily contain a specific antigenic epitope from the pathogen of interest; therefore, they do not contain components of the pathogens that could cause harm when introduced to a host. This means that risks like virulence recovery or incomplete inactivation and pre-existing immunity are significantly eliminated.9 Subunit vaccines are known to cause less adverse reactions compared to inactivated or live vaccines; thus, they are generally safe and are believed to be able to target specific, well-defined neutralising antibodies with enhanced immunogenic efficacy.10 However, due to their purification process, they may be less immunogenic and may also lose their innate immune properties because of reduced antigenic components.11 To deal with these disadvantages, subunit vaccines are usually linked to adjuvants to increase their immunogenic potential.
In this study, the authors try to propose a subunit vaccine by identifying a vaccinable complex that comprises components from CHIKV structural proteins with antigenic potentials. The viral antigens selected for the study include 6K, E1, and Cp proteins, which are known parts of CHIKV structural protein,12,13 and play vital roles in the virus pathogenicity.
CHIKV Cps are involved in the virus RNA binding and particle assembly after replication. The Cp is a small-sized monomeric peptide of approximately 30 kDa, and as such, it can passively transport through nuclear pores. There are reports of Cp directly interacting with host cell transport receptors such as the CRM1 receptors to transport CHIKV proteins.14. The Cps are also known antigens capable of inducing the production of Cp-specific antibodies within the host.14
E1 proteins are part of the envelope protein E1-E2 heterodimer triplets that form the spike glycoprotein found on the surface of CHIKV. CHIKV uses the E1-E2 heterodimer to interact with several host receptors, such as the glycosaminoglycans and Mxra8, to achieve cell attachment and entry.15 Once E1-E2 heterodimer attachment is complete, pH-dependent membrane fusion and endocytosis mediated by clathrin bring about the intake of the particle. The low pH of the host cell causes the E1-E2 heterodimer to dissociate, thereby exposing the fusion residues located at the E1 cd loop. The fusion residues then bind to the target receptor, which causes E1 to conform by back folding upon itself and trimerising, resulting in viral entry.16
Alphavirus 6K proteins are small peptides, typically 58–61 amino acids long. They are hydrophobic and membrane-linked. These proteins act as a stop-transfer sequence for the insertion of E1 proteins into the endoplasmic reticulum membrane. 6K protein expression also boosts membrane permeability and viral budding in the cells of eukaryotes. The alphavirus 6K protein is suggested to be a viroporin (a viral ion channel) due to its characteristic features, such as being an integral membrane protein, its short length (50–120 amino acids long), its role in virus assembly and release, and its function in membrane permeability of infected cells.17
The structural glycoprotein E1 has been reported to cause neutralising antibody production by the host cells, which are capable of clearing CHIKV and thus protect vaccinated hosts. E1 glycoprotein is reported to be more antigenic when linked with an adjuvant.8 It has been used in vaccinating experimental animals along with other immune-inducing glycoproteins, such as E2 glycoprotein, in combination with adjuvants. The results obtained show that vaccinable complexes, like those developed from glycoproteins such as E1 and E2, can actively neutralise CHIKV.18
There is very little information on the application of chikungunya Cps and 6K glycoprotein in the development of a vaccine against the virus, but they were selected in this in silico study due to their high antigenic score on the antigenic prediction tools ANTIGENpro.19
With the selected CHIKV peptides E1, 6K, and Cps, the use of in silico strategies was focused on identifying epitopes within the conserved regions of these immune-stimulating peptides that can be recognised by B cells, as well as being able to bind to toll-like receptors during host immune recognition for the development of a possible subunit vaccine. This was achieved by first retrieving the peptides of interest from available databases; confirming their antigenic propensity on antigen-predicting tools; determining the conserved regions within the selected peptides; confirming the antigenic potential of the selected conserved regions; and determining the presence of antigenic epitopes that can sensitise B cells, cytotoxic T lymphocyte (CTL), and helper T lymphocyte (HTL) production. Next up was constructing a primary structure of the possible vaccine by linking the CTL, HTL, and B cell antigenic epitopes with appropriate linkers; predicting the possible secondary and tertiary structure of the proposed vaccine; validating tertiary structure; and finally, docking the proposed vaccine with a toll-like receptor 4 (TLR4).
METHODOLOGY
Retrieval of Chikungunya Virus Structural Protein Sequences From UniprotKB
FASTA format sequences of CHIKV structural proteins were downloaded from the UniprotKB database (National Institutes of Health, Bethesda, Maryland, USA),20,21 which is a curated database that acts as a central access point for integrated protein information. The database provides a comprehensive compendium of all reported protein sequences obtained from laboratory experiments or computational predictions. The structural proteins retrieved for this research were Cp proteins, 6K glycoproteins, and E1 glycoproteins. The selections of these proteins was informed on the basis of their antigenic properties, as previously explained.22, 23
Multiple Sequence Alignment
Selected sequences were aligned on the multiple sequence alignment tool using the ClustalW algorithm on the MEGA X software (Pennsylvania State University, State College, USA). MEGA X carries out statistical analysis for molecular evolution. The alignment explorer of MEGA X is a built-in tool for viewing, editing, and creating alignments through the ClustalW and MUSCLE programs to identify conserved regions common among sequences. The ClustalW algorithm globally aligns the strings of the sequence and compares the whole length of the sequences with each other to identify rejoins of similarity and identity.13,14 Identifying the conserved regions within the selected proteins is important for the identification of a universal vaccine capable of inducing immunological response for all variants of the virus.6
Antigenic Screening of Conserved Epitopes
The conserved sequences, selected after multiple alignments, were subjected to antigenic screening on the ANTIGENpro server.19 This server is a sequence-based prediction tool capable of predicting the antigenicity of whole proteins and providing the probability score for the predicted antigenicity. The tool is trained with reactivity data obtained by protein microarray analysis of five pathogens. Epitopes with an antigenic score of 0.8, which is the cut-off mark for antigenicity on the ANTIGENpro, are accepted as immunogens.24
Screening for Cytotoxic T Lymphocytes From Conserved Epitopes
Since antigens bind to major histocompatibility complex (MHC) Class I molecules to initiate immune responses, the conserved epitopes that passed antigenic screening were analysed on the NetCTL version 1.2 web server25 to confirm their CTL activating potential. NetCTL version 1.2 web server is a human CTL epitope prediction tool that integrates MHC Class I affinity, proteasomal cleavages, and transporter associated with antigen processing efficiency predictions to identify human CTL epitopes.26
Screening for Helper T Lymphocytes from Conserved Epitopes
While CTL epitopes initiate an immune response by binding to the molecule of MHC Class I, MHCs Class II initiate an immune response and memory cell formation by presenting surface antigens to T cells and B cells. Researchers used the Immune Epitope Database (IEDB)27 to predict the HTL epitopes in the conserved sequences of CHIKV structural protein.28
Screening for B Cells from Conserved Epitopes
Another immune-stimulating epitope of interest, which was predicted on the BCPred web server29 from the conserved sequences, is the B cell epitope. B cell epitopes are essential for peptide-based vaccines, as they recognise and bind to B cell epitopes to initiate an immune response and memory cell, which is a key component to initiating an immune response. At the default setting, epitopes with a score of 1 are selected as B cell epitopes.30
Constructing Possible Primary Structure of Proposed Vaccine
A potential primary vaccine construct was developed by linking the CTL, HTL, and B cell epitopes with appropriate linkers, such as Ala-Ala-Tyr (AAY), which was chosen on the basis of its function. The AAY linker is the mammalian cell proteasome cleavage site, known to increase immunogenicity of multi-epitope vaccines. Epitopes linked with AAY result in effective separation within the cell, thereby reducing junctional immunogenicity. HTL and B cell epitopes were also linked by GPGPG, which has been shown to induce HTL responses, an important multi-epitope vaccine property. Like AAY, it is also able to prevent junctional immunogenicity.
Following the respective linking of HTL and B cell epitopes, the linked CTL, HTL, and B cell epitopes were further joined to each other to form a long chain of polypeptide by the GPGPG linker in the format: [CTLAAYCTL]nGPGPG[HTLGPGPGHTL]nGPGPG[B-cellsGPGPGB-cells]n.
To complete the construction of the primary vaccine structure, the team attached an adjuvant with the linker EAAAK to the N-terminal of the long chain of polypeptides, which were linked together by AAY and GPGPG linkers. This is because, for a multi-epitope vaccine to be strongly immunogenic, adjuvants are required, as they catalyse innate immune responses by inducing a localised inflammatory reaction initiated by interactions between TLRs and antigens. However, most synthetic adjuvants, such as aluminium-based adjuvants, result in adverse effects. So, in an effort to mitigate these side effects, natural candidates like the TLR agonist (RS09) were developed. In this study, RS09 was selected as the adjuvant of choice, as it is capable of stimulating and activating TLR4 and NF-kB, which is reported to result in an increased antibody production.31 The EAAAK linker used is a solid α-helix-forming protein, which contains intra-molecular hydrogen bonds and a closely packed backbone. EAAAK maintains a fixed distance and, hence, reduces interference between the linked epitopes. This ensures that the epitopes keep their individual functionality within the functional domain. With this, the final primary construct of the vaccine now has a structural format: RS09EAAAK[CTLAAYCTL]nGPGPG[HTLGPGPGHTL]nGPGPG[B-cellsGPGPGB-cells].32
Scoring the Allergenic and Antigenic Potential of the Primary Construct and Predicting its Physicochemical Properties
Allergenic screening
Vaccines are designed to be an immunogen without causing allergic reactions. The primary structure of this vaccinable complex was assessed on the AllerTOP v.2.0 server33 to confirm its non-allergenic properties. AllerTOP v.2.0 predicts allergenicity by using the K nearest neighbours (KNN) model, and is reported to have an 85.9% accuracy.34
Antigenic screening
Predicting the antigenicity of the vaccinable complex was achieved on the ANTIGENpro server: a complex with an antigenic score ≥0.8 is considered an antigen. To confirm the antigenic potential of the complex after ANTIGENpro prediction, the complex was subjected to the VaxiJen online server set at a threshold of 0.4 and selecting virus as the source of the complex. A score ≥0.4 is considered an antigen and confirms the antigenicity of the vaccine complex.
Prediction of the physicochemical properties of the primary construct
Predicting the physicochemical properties of the primary construct is indicative of the physicochemical behaviour of the complex. Predicting the physicochemical parameters involves predicting properties of the complex, such as the grand average of hydropathy (GRAVY) score, isoelectric point, instability index, aliphatic index, etc. Physicochemical prediction of the vaccine primary structure was done on the ProtParam web server.35
Secondary structure prediction
Secondary structure of a polypeptide refers to the spatial arrangement of said polypeptide, consisting of α-helices, β-sheets, and turns. Predicting the secondary structure of a vaccine gives information on how the vaccine polypeptides are folded, and assists in determining how the vaccine will interact with the host immune system, such as whether the vaccine possesses the right conformation. Predicting secondary structure affords a chance to optimise the vaccine to achieve stability and enhanced antigenicity for an efficacious vaccine. The secondary structure of the vaccinable complex was predicted on the Self Optimized Prediction Method (SOPMA) web server.36 This web server is an improved secondary structure prediction tool capable of predicting secondary structure with a higher success rate. SOPMA has been reported to have 69.5% accuracy for predicting the α-helix, β-sheet, and coils of protein secondary structure in a database holding 126 chains of non-homologous polypeptides.34,37
Tertiary structure prediction
The 3D model of the proposed vaccinable complex was predicted on the I-TASSER server.38 The server predicts the 3D structure of polypeptides by assessing quantitative scores. The server provides five outputs of full length for every query input, along with other parameters, such as standard deviation, TM-scores, confidence score, and root mean square deviation.39 I-TASSER was selected as the best server for protein structure and function prediction. It predicts proteins by first identifying structure templates from Protein Data Base (PDB) using multiple threading methods and full-length atomic models built by iterative template-based fragments.
Linear and Discontinuous B Cell Conformational Epitopes Mapping
B cell epitopes are comprised of either linear (continuous) or conformational (discontinuous) epitopes. Linear B cell epitopes are sequential epitopes and make up a lesser percentage of the B cells, while conformational B cell epitopes are composed of fragments of solvent-exposed non-sequential epitopes, which make up the larger percentage of B cells. It becomes important to map both the linear and discontinuous B cell epitopes, because antibodies that recognise linear B cell epitopes may not be able to recognise conformational B cell epitopes. To map the B cell epitopes of vaccine constructs, Ellipro suite is used.40 The web tool is set at a minimum threshold of 0.5, and a maximum distance of 6. Ellipro suite uses both the MODELLER program and Jmol viewer to evaluate and visualise the 3D structure of B cell epitopes of vaccine constructs.24
Refinement of the Predicted Tertiary Structure
The tertiary structure of a protein is dependent on the peptides it is composed of; both the peptide composition and structure of the protein determine the protein’s function. If a protein does not conform to the native structure, it may lose its natural function. To ensure that the 3D model of the study’s predicted protein is as structurally similar as possible to the native protein structure to which it was modelled, refinement needs to be done. 3D structure refinement of the vaccine candidate was done on the GalaxyWEB server.41 Refinement increases the number of residues in the favoured region. GalaxyRefine uses CASP 10 and functions by reconstructing the protein side chains, relaxing the structure by repacking, and molecular dynamics simulation.34
Tertiary Structure Validation
Once refinement was done on the authors’ vaccinable complex, they validated the refined structure on the ProSA web tool,42 which is used for checking potential errors in the 3D model of experimentally determined protein structures, theoretical models, and engineered protein. Validation was obtained as Z-score and local model quality. PROCHEK was also used for the Ramachandran plot analysis, which validates and verifies the complex functionality.24
Molecular Docking of Vaccinable Complex With TLR4
Visually studying the interaction of a ligand-receptor bonding, like that of an antigen-antibody interaction, requires an in silico simulation of the interaction. The authors studied the interaction of their complex with TLR4, the tool ClusPro 2.0 server.43 ClusPro 2.0 predicted the binding domain between the vaccine construct and TLR4 in their most stable complex form and lowest binding energy.44
RESULTS
Retrieval, Multiple Sequence Alignment, and Antigenic Screen of the Conserved Epitopes of CHIKV Structural Protein Sequences
A total of 125 CHIKV structural protein sequences, comprising 13 Cps, 22 6K glycoproteins, and 90 E1 glycoproteins, were retrieved from UniprotKB database in FASTA format. The retrieved sequences were analysed to identify epitopes of shared conserved regions. These epitopes were selected and screened for their antigenic properties, which revealed that all the conserved epitopes were antigenic as they had antigenic scores ≥0.8.
Screening for Cytotoxic T Lymphocytes, Helper T Lymphocytes, and B Cells From the Conserved Epitopes
Seventeen 9mers CTL epitopes with high comb scores were selected among the predicted epitopes from the conserved CHIKV structural epitopes. A total of 32 15mers HTL epitopes were also selected from the total predicted HTL epitopes on the basis of their low percentile rank score, and 16 B cell epitopes were selected from the total B cell epitopes predicted from the virus structural protein based on their high score. These are shown in Table 1.
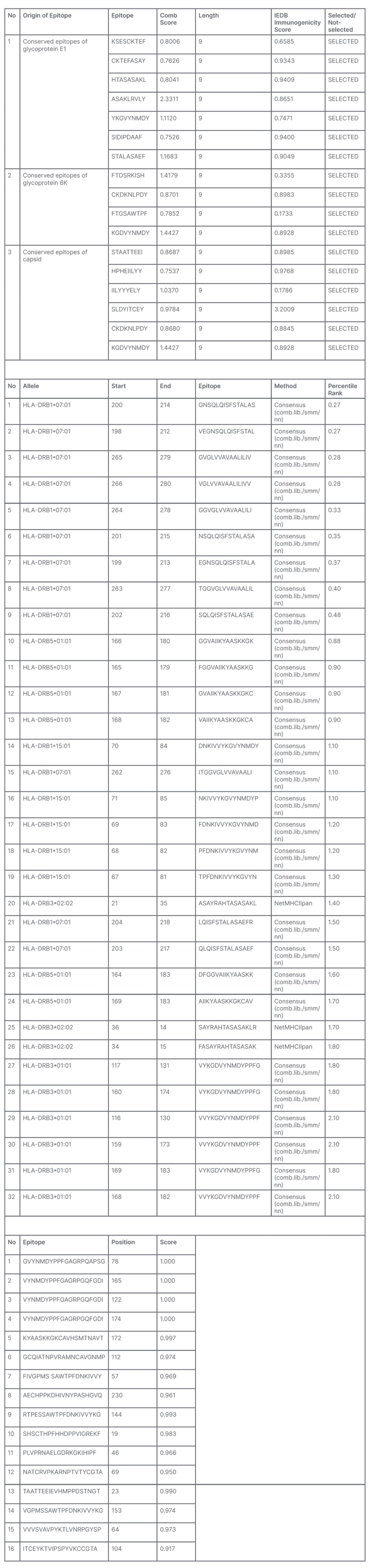
Table 1: Predicted cytotoxic T lymphocytes, helper T lymphocytes, and B cells.
IEDB: Immune Epitope Database.
Construction of Primary Vaccine Candidate
A total of 1,236 amino acid residues, comprising the selected CTL, HTL, and B cells linked with appropriate linkers, were used to construct the primary vaccine candidate. An adjuvant was linked to the N-terminal of the vaccine candidate to improve its antigenicity. Figure 1A shows a schematic presentation of the primary vaccine structure.
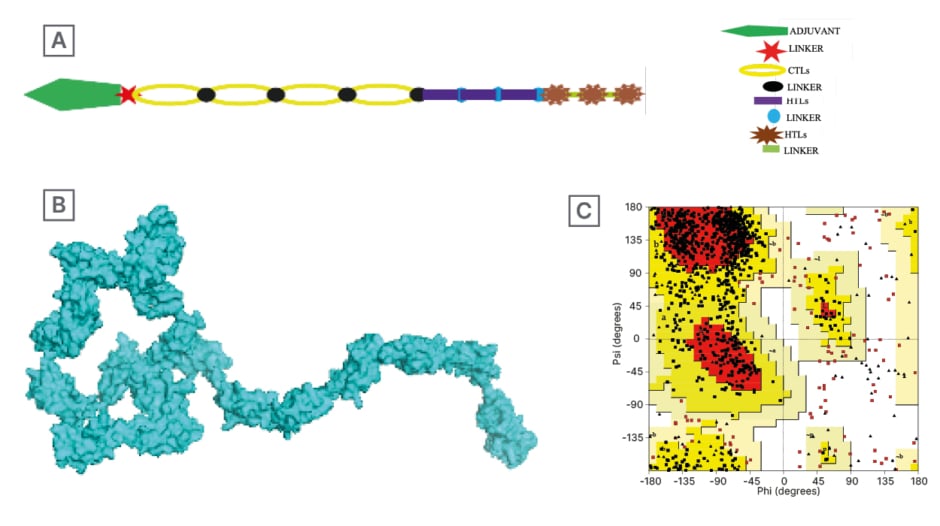
Figure 1: A) Schematic representation of the primary vaccine structure; B) Predicted tertiary structure of vaccine candidate; and C) Ramachandran plot analysis for the candidate vaccine.
The Ramachandran plot analysis for the candidate vaccine shows that 97.2% of the residue is in the most favoured region, 2.8% of the residue is in the allowed region, and 0.0% in the disallowed region.
CTL: cytotoxic T lymphocyte; HTL: helper T lymphocyte.
Assessment of Allergenic, Antigenic, and Physicochemical Properties of the Vaccine Primary Construct
The primary vaccine construct was predicted to be non-allergenic on the allergen predicting tool AllerTOP v.2.0. It was also confirmed to be antigenic with a high score of 0.946655 on the ANTIGENpro server; and a score of 0.4621 on the VaxiJen online server confirmed the antigenicity of the proposed vaccine, indicating it to be antigen with potential to induce immune response.15 Physicochemical assessment of the primary structure revealed that the sequence has a molecular weight of 125060.86, instability index of 23.96, aliphatic index of 65.11, and GRAVY of -0.121.
Vaccine Secondary Structure Prediction
Secondary structure prediction of the vaccine candidate on the SOPMA server predicts the composition of the secondary structure of the vaccine candidate. SOPMA predicted a 17.96% α-helix, 27.67% extended strand, 49.68% random coil, and 4.69% β-turns for the vaccine construct.
Vaccine Tertiary Structure Prediction
Predicting tertiary structure of the vaccine candidate was carried out on the I-TASSER server. Five models were predicted as having favourable properties, but Model 1 was selected on the basis of its confidential score of 0.59, estimated TM-score of 0.79±0.09, and root mean square deviation of 8.0±4.4Å. The 3D structure is shown in Figure 1B.
Linear and Discontinuous B Cell Conformational Epitopes Mapping
Ellipro suite predicted a total of 244 residues for the linear B cell epitopes, which were between residue numbers 993–1,236; the predicted score for the linear B cell epitope was 0.855. A 206 residue for discontinuous B cell epitope was also predicted for the vaccine construct with a score of 0.856. The linear and discontinuous B cell epitopes were visualised with the Jmol viewer, as seen on Figure 2.
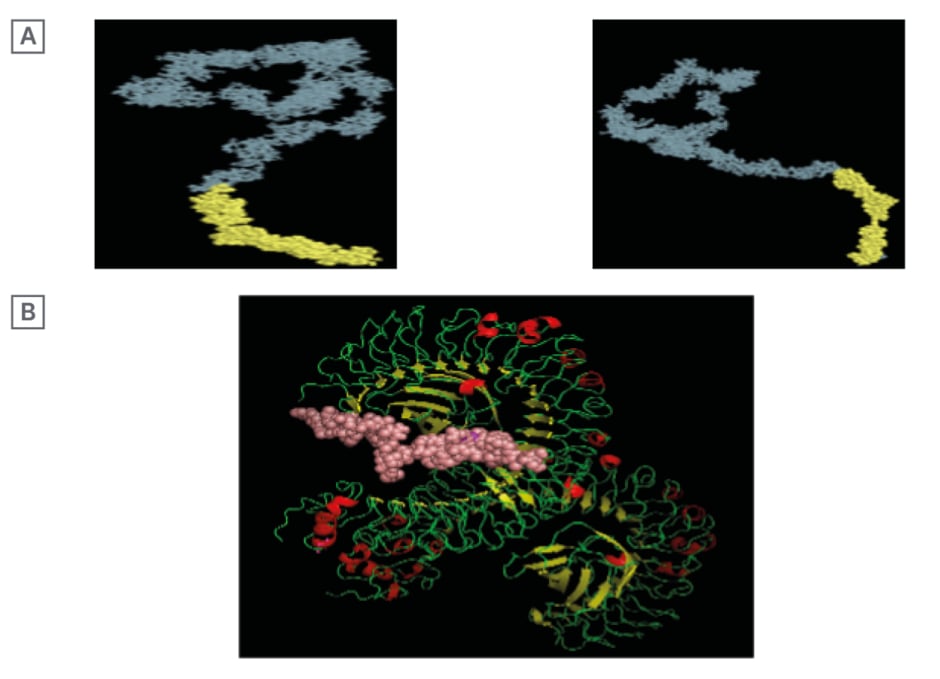
Figure 2: A) Conformational linear B cell structure (yellow) attached to the vaccine structure (grey-blue); and B) 3D representation of docked vaccine candidate with toll-like receptor 4.
Refinement of Tertiary Structure Prediction
The refinement of the vaccine tertiary structure provided five models; Model 1 (Figure 1B) was selected as the best model.
Tertiary Structure Validation
Validating the final structure of the vaccine candidate requires that the PDB file of the vaccine be subjected to a few statistical validation softwares. The result obtained revealed the vaccine’s structure Z-score, graph of the vaccine local model quality, and Ramachandran plot analysis. These are shown in Figures 1C and 1D.
Molecular Docking of Vaccine Construct with TLR4
In silico molecular docking of the vaccine candidate with TLR4 on the ClusPro server predicted 10 possible models at the balance setting. Model 0 was selected based on the binding energy (-1,072.8). This is shown in Figure 1.
DISCUSSION
Outbreaks of CHIKF have been reported over the years across many countries within Africa, Asia, and Polynesia. The largest outbreak of the disease on record was in 2004–2007 in the Indian Ocean Island and India, where a serological survey suggests that more than 60% of the population in the region was exposed to the virus.32 The first recorded outbreak of the disease occurred in the Newalla and Masisi districts in the southern province of former Tanganyika Territory, now Tanzania, between 1952–1953, with a 23% incidence rate.46,47 Since the first outbreak, the disease has spread from East Africa to Asia and Europe.
For an infectious disease with no known treatment and high potential to spread, the most realistic means to manage it is preventive measures; only with prevention can there be a chance for control.
This research focuses on identifying viral epitopes with potential for vaccine development. In this study, CHIVK structural proteins capable of causing sensitisation of the immune system were selected, and conserved epitopes from these selected protein were analysed for their CTL, HTL, and B cell recognition.8, 18
To determine the conserved regions of the E1, 6K, and Cp proteins, each protein type was respectively aligned on the Mega X tool, and their conserved regions identified. Identified regions were selected for further analysis. Identifying conserved regions ensures that only regions within sequences shared among all available variants of CHIKV are selected and used for vaccine design. Using conserved regions makes sure that all variants of the virus found on the database at the time of this research were represented, guaranteeing the vaccine efficacy to confer protection against all variants of CHIKV.48 Nevertheless, using conserved regions for vaccine design comes with its drawbacks. One of the most important challenges of using conserved regions for vaccines is the risk of altered immunogenic potential of the sequence. When this happens, it is usually a result of parts, or all, of the active sites being lost because they are located in the region identified as non-conserved. To ensure that the selected conserved sequences maintain their antigenic potential, the sequences should be screened for antigenicity.
In this study, the researchers screened for the antigenicity of the conserved regions of the selected epitopes using the ANTIGENpro online tool. All of the authors’ selections were predicted to have antigen scores >0.8 (Table 1). These are good indications of their immune stimulating properties, which make them promising epitopes for vaccine development against CHIKV infection considering that the antigen of CHIKV is limited and lifelong immunity usually occurs after infection.18 Hence, vaccinating a host with this candidate vaccine is expected to produce similar results.
After constructing the primary structure of the vaccinable complex, researchers identified epitopes that can be recognised by HTLs, CTLs, and B cells. Identification of HTL epitopes is an important step in in silico vaccine development, because HTLs are responsible for activating CTLs, B cells, and macrophages in adaptive immunity.49 The team predicted CHIKV epitopes recognisable by HTLs on the IEDB platform.27 The tool predicted 32 15mers HTL epitopes from the E1, 6k, and Cp protein. CLT and B cell epitopes were predicted, as earlier stated, to identify epitopes that can initiate cellular and humoral immunity. This is shown on Table 1. Predicting these epitopes was important because vaccines are required to induce antibodies and memory cell production.50 Mahmoodi et al.51 and Zakaria et al.52 reported to have identified CT, HTL, and B cell epitopes from CHIKV structural protein. In their research, Mahmoodi et al.51 reported that the CTL, HTL, and B cell epitopes identified showed high immunogenic potentials and conferred immunity against CHIKV infection. Zakaria et al.52 also reported the activation of both humoral and adaptive immune cells, and production of various cytokines by the structural epitopes capable of controlling CHIKV. On the premise of the reports of these two authors, the team selected epitopes of CHIKV structural protein with high antigenic scores that qualified them as an active component for an effective CHIKV multi-epitope vaccine candidate.51,52
An active multi-epitope vaccine should contain all the epitopes that can induce all various immune cells. In this research, the immune cells of interest are the HTL, CTL, and B cells. The epitopes with the potential to induce these cells were predicted and are reported in Table 1. Designing a vaccine using the in silico method requires the predicted epitopes to be linked to form the primary vaccine structure. To achieve this, the HTL epitopes were linked by GPGPG. AAY was used to link CTLs together, and B cells were also linked together with GPGPG. The linked epitopes were further linked by GPGPG, with the HTLs at the N-terminal and the B cells at the C-terminal. At the N-terminal, an adjuvant is connected to the vaccine polypeptide chain by a specific linker, EAAAAK, to create a primary vaccine structure: RS09EAAAK[CTLAAYCTL]nGPGPG[HTLGPGPGHTL]nGPGPG[B-cellsGPGPGB-cells (Figure 1).
The primary structure of the vaccinable complex was further assessed to determine its allergenic propensity, antigenic potential, and physicochemical properties. It was found that the vaccine molecule is a non-allergen with a high antigenic potential, scoring an antigenic score of 0.946655 on the ANTIGENpro server and a score of 0.4621 on the VaxiJen server. Owing to the molecular weight (125060.86 Da) of the vaccine complex, it is confirmed that the vaccine consists of multiple antigenic regions, which makes it capable of stimulating a robust immune response. Its instability index score was 23.96; this meets the <40 threshold, indicating the vaccine’s structural stability. The aliphatic index of 65.11 also provides information on the vaccine’s thermo-stability. The negative GRAVY score of the vaccine molecule indicates its hydrophilic nature, providing information on its water-soluble property. A similar approach was used by Foroutan et al.53 to develop a vaccine candidate against Toxoplasma gondii. Reporting results with comparable physicochemical properties to these, Forountan et al.53 went further to validate their vaccine by carrying out animal trials using laboratory mice. The team reported that their vaccine induced a strong humoral and cellular immune response in the mice. Findings corresponding to this were also reported by Sumera et al.54 in their publication in 2022. By comparing results with the results from these, and other papers, it was established that this vaccine’s primary structure physicochemical properties were within an accepted threshold.55
The secondary and tertiary structure of the vaccine were predicted following the construction of the primary structure. These were done on the SOPMA and I-TASSER servers, respectively. The linear B cell epitopes were also predicted before refinement of the tertiary vaccinable complex was done on the Galaxy refine server. Refinement is necessary to ensure the vaccine tertiary structure conforms to the native protein.
The team structurally validated the vaccinable complex to ensure that the sequence and structure used behave like those of native proteins from which it is predicted. From the structural validation analysis, an overall model quality graph and Z-score (–1.52) were obtained, which validate the vaccine tertiary structure as conforming to native structure. The graph of the Z-score revealed that the protein structure is determined by NMR spectroscopy (Figure 1C). The Z-score value and the contour line under the 0.0 threshold of the overall model quality graph both indicate the good quality of the predicted candidate vaccine tertiary structure. This is because the authors’ tertiary structure has very little 3D structural error. Ramachandran analysis carried out revealed that the structure possesses proteins with 97% of its residue in the favoured region, while none of the residue was in the disallowed region. All of these data validate the quality of this vaccine as a potential vaccine candidate.
TLR4 is expressed in various immune cells in humans, such as macrophages, monocytes, and granulocytes. TLR4 binding prediction is vital in vaccine design and development because TLR4s recognise pathogen-associated molecular patterns and induce immune response. Activating TLR4 improves antigen presentation, adaptive immune response, cytokine production, and vaccine effectiveness.56 It is because of these functions of TLR4, to both innate and adaptive immune activation, that a TLR4 agonist such as RSO9 is used as adjuvant in epitope vaccines to enhance their immunogenicity.57 To evaluate the binding affinity of the candidate vaccine tertiary structure to the antigen recognising receptors, a TLR4 molecule was docked with the PDB structure of the vaccine candidate on the Cluspro server. Ten models were predicted, and Model 0 was selected on the basis of its low binding energy of -1,072.8. Low binding energy is significant in vaccine design and development, as it is indicative of the strength of the protein-ligand binding complex. The lower the binding energy, the stronger the complex, hence, the more robust the immune response. Binding energy also gives insights into the structure of the protein-ligand complex. With that information, decisions for more effective vaccines can be made. From this study, the low binding energy obtained for Model 0 informed the selection. Model 0 is predicted to have the strongest and most stable binding interaction. The structure of the docked complex is shown on Figure 2.
Multi-epitope vaccines against CHIKV are predicted to be advantageous in many ways, including broader protection against diverse strains of the virus, capturing the risk of escape mutant by enhanced cross-reactivity;58 and reducing host immune evasion, their potential for long-lasting immunity, their flexibility in design, and their higher level of safety. Regardless, developing a multi-epitope vaccine for CHIKV poses several challenges and limitations, such as the challenge of antigenic diversity of CHIKV and the possibility of low immunogenicity of some of the epitopes. T cell response, which has always been an important part of multi-epitope vaccine design, and the long-term protection they confer on the host, is also a vital research area for improving multi-epitope vaccines.59 To correct these challenges, a CHIKV vaccine must consist of various epitopes for it to have the immune coverage that accounts for all the diversity of CHIKV. It is also imperative to identify immunologically viable epitopes to use as component for a multi-epitope vaccine.60 Attaching adjuvants to the CHIKV vaccine is an important process in its design and development; this is because it greatly improves the immunogenicity of the vaccine. In the in silico design and development of this vaccine candidate, the adjuvant RS09 was attached to the N-terminal to improve its immune-stimulating properties. There are many known adjuvants, such as the RS09 and CpG oligodeoxynucleotides, that could be used to improve CHIKV vaccine effectiveness. Selecting an adjuvant with maximum efficiency among all the available adjuvants, and developing new and novel adjuvants, should be another area of research that is focused on.61 Apart from deep adjuvant reach, optimising T cell activation by CHIKV multi-epitope vaccines should also be a vital point in future CHIKV multi-epitope vaccine investigations.62 Clinical trials are also necessary step in vaccine development. This is vital in order to validate the safety, effectiveness, and durability of CHIKV multi-epitope vaccines in diverse populations, especially those in endemic regions.
CONCLUSION
The growing scourge of diseases across the globe, including CHIKV, requires immediate response. Preventive medicine has proven to be the most effective strategy for controlling infectious diseases, especially when performed properly. CHIKV, which was first discovered in the United Republic of Tanzania in 1952, has spread to become a global threat and there is no approved treatment for the infection. The symptoms of CHIKF are self-limiting, but the risk of eye, neurological, and heart complications; severe joint pain; or even death is ever looming, resulting in the need for a means to control infection rate.
This study focused on designing a multi-epitope vaccine capable of reducing the spread of CHIKV infection by sensitising the immune system to the infection. This in silico vaccine construct was built from antigenic epitopes from CHIKV 6K, E1, and Cp proteins. The proteins were predicted to be able to activate CTLs, HTLs, and B cells, and bind effectively to TLR4, which is indicative of an efficacious vaccine candidate. The primary, secondary, and tertiary structures were predicted to be composed of structurally stable molecules that interact to create a thermal and chemically durable compound. This was validated by physicochemical and statistical analyses done on various online servers. In conclusion, the candidate vaccine is expected to be effective and capable of inducing antibody production by way of sensitising the host’s humoral and cell-mediated immunity against future exposure to CHIKV. The vaccine candidate would be capable of improving the health of people, especially those in endemic communities, as the vaccine was designed with conserved regions from CHIKV antigenic epitopes, granting it a wide coverage for many variants of the CHIKV. The vaccine being a multi-epitope is safe, as issues such as virulence recovery or incomplete inactivation and pre-existing immunity are eliminated. From the data gathered, the researchers could make an educated suggestion that a vaccine developed using E1, 6K, and Cp proteins would be a promising vaccine that could be effective in the control of the spread of CHIKF. The method applied in this research provides necessary data indicating that the candidate vaccine will be effective in inducing immunological response of the host immune system, inducing production of B cells that can be stored as memory cells and bind perfectly to antigenic receptors of the immune system.