BACKGROUND AND AIMS
Spontaneous renal artery dissection (SRAD) is a rare and often unrecognised clinical entity, which accounts for 1–2% of all arterial dissections. It can be the result of several underlying diseases, such as atherosclerosis, malignant hypertension, fibromuscular dysplasia, and connective tissue disorders.1 Even though atherosclerosis is considered the principal cause of SRAD cases, the aetiology of the majority of cases remains unknown.
There are three different clinical manifestations: sub-acute state without apparent progression; renal infarction state due to an acute occlusion; and chronic state with renovascular hypertension. Renal infarction due to SRAD is often misdiagnosed as acute pyelonephritis, which can cause delays in the diagnosis and treatment.
MATERIALS AND METHODS
The authors discuss five patients with renal infarction due to SRAD who were admitted to their unit in the prior 1.5 years. There were three males and two females, with a mean age of 50±7.8 years.
From the onset, all patients presented sudden-onset flank pain and new-onset hypertension, and were referred to the authors’ unit at least 1 week later. At admission, renal function was normal for all patients, but two presented microhaematuria. Blood tests showed leukocytosis, high C-reactive protein and fibrinogen levels, and hyperhomocysteinaemia.
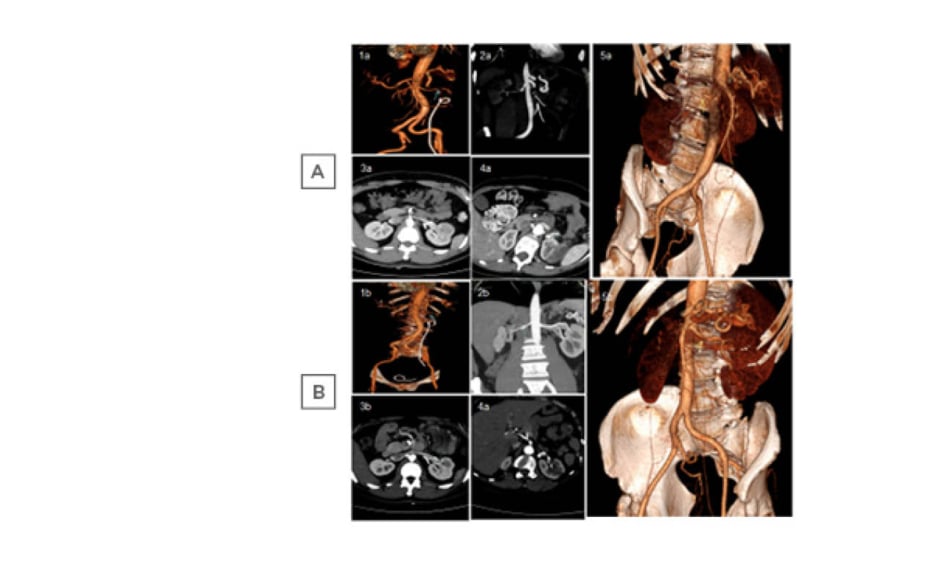
Two males had a family history of cardiovascular disease. Moreover, one was diagnosed with a gastrointestinal stromal tumour, and the other was diagnosed as OFP ostium II. The third male had family history of malignancy. One female had a history of kidney stones and the other had dolichocolon. Abdominal CT angiography (CTA) showed renal infarction due to SRAD, which was bilateral in two patients (Figure 1A).
All patients were treated with anti-hypertensive drugs and systemic anticoagulation (two with continuous intravenous heparin infusion and three with subcutaneous low-molecular-weight heparin), followed by oral warfarin.
RESULTS
At the 3-month follow-up, all the patients had become normotensive and partial or total renal artery recanalisation was found (Figure 1B). All patients had a work-up for hypercoagulable state (activated protein-C resistance, protein S, antithrombin III, antiphospholipids and anticardiolipin antibodies, lupus anticoagulant, and Factor II and V mutation) and for immunological disorders (anti-nuclear antibody, anti-neutrophil cytoplasmic antibodies, complement levels, rheumatoid factor, Ig), all of which were within normal limits or negative, except in one patient who presented a Factor II mutation.
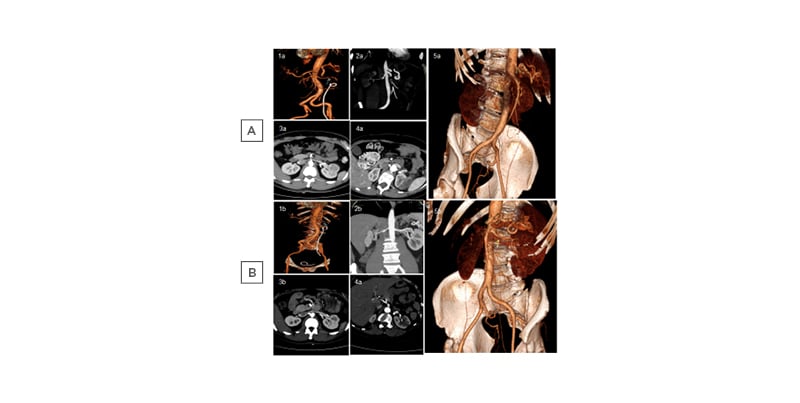
Figure 1: Spontaneous renal artery dissection in five patients at A) onset and B) after 3 months of anticoagulant therapy.
Molecular genetic testing for hereditary aortic diseases and related disorders of connective tissue were performed; one patient was negative, the patient with Factor II mutation showed COL5A1 c.514G>T (heterozygous) mutation, and testing in the three remaining patients is still ongoing.
In literature, over 200 SRAD cases have been reported: one-quarter on autopsy, mostly males (4:1 ratio) in their late 40s to early 50s, usually without any underlying disease, and with bilateral involvement in 10–15% of cases.2 SRAD was most commonly diagnosed by CTA or magnetic resonance angiography. Invasive exploration with angiography is only recommended at an early stage to determine the extent of the lesion and whether endovascular treatment is feasible. The efficacy of oral anticoagulation and anti-hypertensive medication is still controversial. Due to late referral to the authors’ unit, invasive exploration with angiography was not performed; abdominal CTA was diagnostic, and conservative medical treatment was effective in all patients.
CONCLUSION
To summarise, renal infarction due to SRAD seems to be more common than expected in young and healthy patients without apparent underlying disease. Non-invasive procedures and 3-month conservative medical treatment are safe and effective, while molecular genetic testing could be helpful to reveal possible predisposing factors.