Abstract
Magnesium is one of the commonly overlooked electrolytes, yet it plays a vital role in many of the processes in the human body. The balance of magnesium can translate into subtle changes in a person’s daily life, causing fatigue and confusion, to extreme cases that can end up causing central nervous system depression, respiratory failure, or cardiac arrhythmias.
It is vital to be familiar with the physiology of magnesium regulation and knowledgeable regarding the causes that can lead to its toxicity to ensure the prevention of the possibly fatal condition.
Magnesium balance can be summarised as the difference between magnesium intake and its excretion. Any factor overwhelming either of the two factors can cause pathological levels of the electrolyte. In addition to learning preventive measures to help patients against effects of magnesium toxicity, it is also important that the medical community trains to be able to treat cases of hypermagnesaemia.
This review assesses the latest advancements in knowledge of magnesium metabolism, examines the case reports of hypermagnesaemia in an attempt to list the causes of magnesium toxicity, and enumerates management advances for the condition.
INTRODUCTION
Magnesium is a chemical element with an atomic number of 12. It is among the top 10 most abundant elements in nature. In the human body, it is the second most prevalent cation intracellularly and the fourth most common cation extracellularly.1 The body contains approximately 24–26 g (1,000 mmol) of magnesium.
Magnesium plays a fundamental role in the body’s metabolic processes and is involved in more than 300 enzymatic reactions.2-5 It serves as a cofactor in ATPase–phosphate transfer reactions,4,6 oxidative phosphorylation processes,7-9 and synthesis of nucleic acids.10,11 It also actively contributes to glycolysis12,13 and lipid and protein metabolism,14 and affects intracellular signalling and cell membrane stabilisation.15
Magnesium is crucial in balancing the effects of calcium in the body and acts by inhibiting calcium-mediated smooth muscle contraction.16,17 This translates into vasodilatory properties and is important for blood pressure maintenance. Magnesium antagonism of calcium is also integral in the nervous tissues, where calcium helps maintain the resting membrane potential. Excess magnesium levels lead to hyperpolarisation of the nervous tissues, indirectly inhibiting acetylcholine receptors and N-methyl-D-aspartate receptor-mediated central nervous system conductivity, resulting in central nervous system depression.18
PHYSIOLOGY OF MAGNESIUM
Sixty percent of the magnesium in the body is stored in the bones. The remainder is distributed between the intracellular compartment of the muscles and soft tissues, with only 1% found in the extracellular compartment.19,23 Ionised magnesium constitutes 64% of the extracellular magnesium. The remainder is attached to albumin and other anions in the blood.19 Magnesium is referred to as the ‘orphan electrolyte’ due to its lack of specific endocrine control. Thus, serum magnesium levels are consistently maintained by a strict balance between intestinal absorption, renal excretion, and bone buffering of magnesium. The skeletal system has the ability to provide 30% of its magnesium stores in settings of hypomagnesaemia.24
Magnesium Intake
Diet is the primary source of magnesium for the body; the latter half of the small intestine contributes to approximately 80% of the dietary magnesium absorption,25 and the rest occurs in the proximal small intestine and the colon (Figure 1). The majority of intestinal magnesium absorption occurs through the paracellular pathway and is driven by electrochemical gradients.26,27 Transcellular absorption also occurs via transient receptor potential channel subfamily M (TRPM) member 6 (TRPM6),28 possibly via TRPM7,29 and through magnesium–anion complex channels.30 The absorption of magnesium from enterocytes to the bloodstream is facilitated by the sodium/magnesium-ATPase, which is found on the basolateral side of the intestinal cells and uptakes a plasma sodium ion into the enterocyte in exchange for a magnesium ion absorbed into the plasma. The intracellular sodium gradient is balanced by sodium/potassium-ATPase (Na+/K+-ATPase) on the basolateral side, which pumps the sodium ions back into the plasma from the intestinal cells, making them available again for the sodium/magnesium exchanger. This sodium exchange, assisted by concentrations of chloride ions, bicarbonate ions, and fatty acids, ensures an electrochemical gradient favouring magnesium uptake into the intestinal cells and then the plasma.
Depending on the magnesium levels, the intestine can absorb 11–65% of oral magnesium content.24 Despite the lack of a specific endocrine control, magnesium is regulated by multiple factors such as parathyroid hormone, thyroid hormone, growth hormone, vitamin D, and dietary sodium content.31 The intestinal mobility and intestinal epithelial health also play a vital role in magnesium homeostasis. Changes in the intestinal transit time and the intestinal blood supply directly correlate with serum magnesium levels. The daily recommended dose of magnesium is 4.5 mg/kg24 and up to 350 mg.32 Magnesium is abundant in dairy products, fruits, vegetables, fish, meat, legumes, and nuts.
Magnesium Elimination
Active magnesium excretion is fundamental in preventing magnesium accumulation. In the body this is mediated by only two organ systems: the kidneys and the skin (via sweating). Even though the intestinal tract does not have an active role in plasma magnesium excretion, it can waste the surplus dietary magnesium by downregulating the intestinal absorption based on serum magnesium levels.33 The kidneys play a pivotal role in maintaining an optimal magnesium concentration in the body. Since 20% of the serum magnesium is albumin bound, a healthy kidney filters 80% of serum magnesium and usually reabsorbs 95% from the filtrate. However, in magnesium-depleted states, the kidneys can increase this reabsorption capacity to 99.5%.34 On the other hand, the kidneys usually excrete approximately 100 mg of magnesium per day in the urine but have the ability to excrete up to 70% of the filtered magnesium (Figure 1).
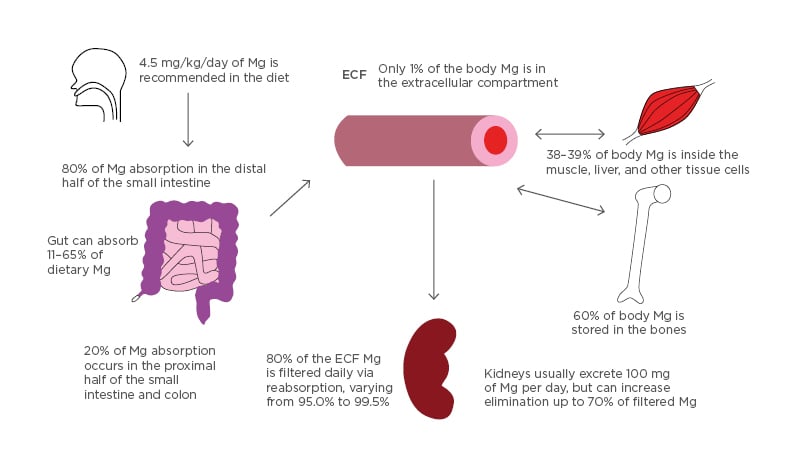
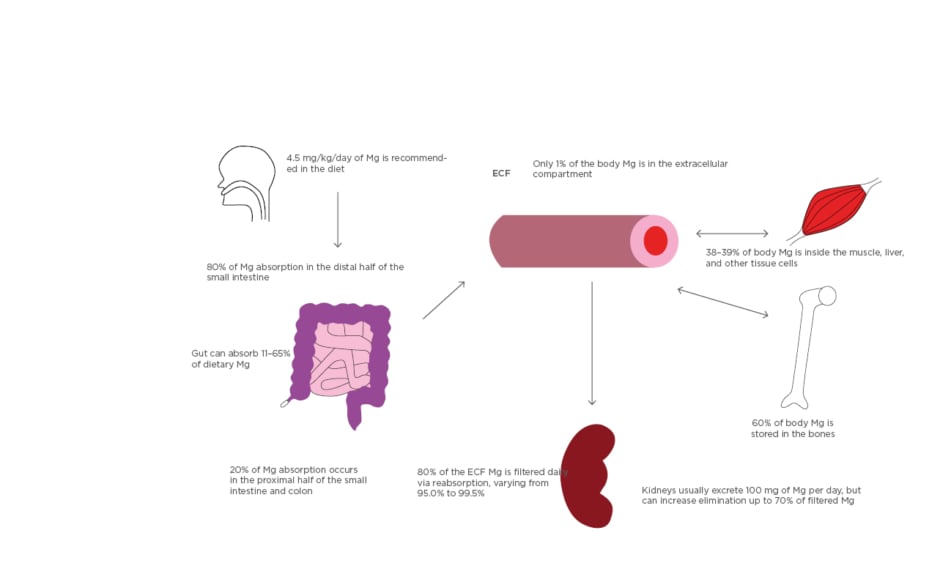
Figure 1: Absorption and distribution of magnesium.
All Mg intake is usually by mouth. 4.5 mg/kg/day of dietary magnesium is recommended. Eighty percent of dietary Mg is absorbed in the later half of the small intestine and the remainder in the rest of the small and large intestine. The gut can vary in the absorption of dietary magnesium content (11–65%). Of the 24–26 g Mg in the body, 60% is in the bones, 38–39% in the liver and muscle tissue, and 1% is in the extracellular compartment. Kidneys usually filter 80% of serum magnesium per day and can vary reabsorption from 95.0–99.5% of the filtrate. Urine Mg content can vary from 0.1 to >1.0 g/day.
ECF: extracellular fluid; Mg: magnesium.
Renal handling of magnesium differs from other electrolytes in that the predominant site of tubular reabsorption is the thick ascending limb (TAL) loop of Henle rather than the proximal convoluted tubule. Indeed, the TAL reabsorbs 70% of the filtered magnesium while approximately 15% is reabsorbed in each of the proximal convoluted tubule and distal convoluted tubule (DCT) (Figure 2).
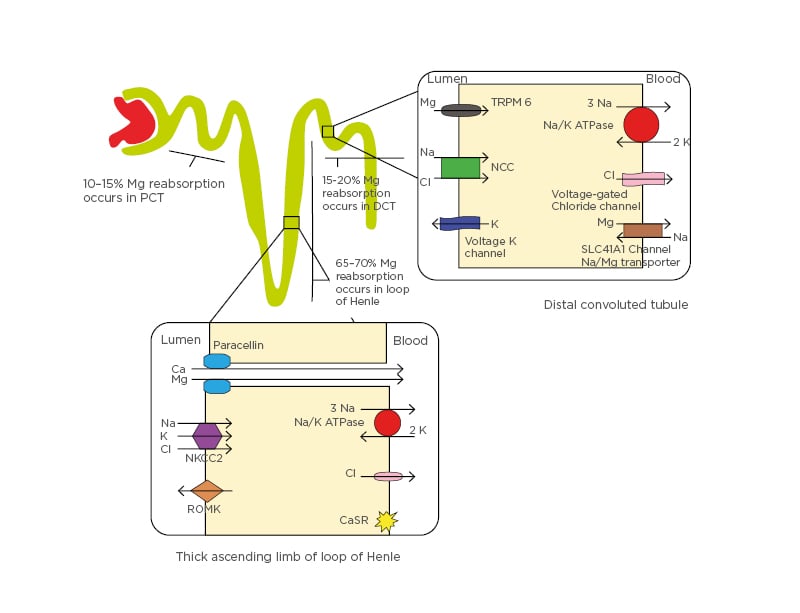
Figure 2: Renal handling of magnesium.
Mg reabsorption occurs 60–70% in the loop of Henle, 10–15% in the PCT, and 15–20% in the DCT. In the loop of Henle, the NKCC2, the renal outer medullary K channel on the luminal side, and Na+/K+-ATPase on the basolateral side maintain a positive gradient in the lumen by secreting K into the channel. This helps create an electrochemical gradient between the lumen and the blood, forcing Ca and Mg reabsorption through paracellular movement. The CaSR on the basolateral side of the loop of Henle cells also influences activity of NA+/K+-ATPase and the NKCC2, which inhibits their activity and controls the electrochemical gradient. In the DCT, Mg reabsorption occurs through the TRPM6 and TRPM7 on the luminal side and the SLC41A1 protein at the basolateral side, which is proposed to act as a Na/Mg counter-transporter. Voltage-gated K channel-guided potassium secretion, the NCC at the luminal side, and voltage-gated Cl channels help control the flow of Mg ions.
Ca: calcium; CaSR: calcium-sensing receptor; Cl: chloride; DCT: distal convoluted tubule; K: potassium; Mg: magnesium; Na: sodium; NCC: sodium chloride cotransporter; NKCC2: sodium-potassium-2 chloride cotransporter; PCT: proximal convoluted tubule; ROMK: renal outer medullary potassium channel; SLC41A1: solute carrier family 41 A1; TRPM6: transient receptor potential channel subfamily M member 6.
Most of the reabsorption in the TAL occurs through paracellular transport with assistance from paracellin-1 membrane proteins, mediated by the electrochemical gradient between the tubular lumen and interstitium. This gradient is established in the TAL by maintaining a net-positive gradient of +8 mV (positive charge) in the tubular lumen via potassium excretion back into the tubule via the renal outer medullary potassium channel (ROMK), after initial potassium influx by sodium-potassium-2 chloride cotransporter (NKCC2).
By regulating and fine-tuning the magnesium reabsorption at the very end, the DCT regulates the amount of magnesium in the urine and is thus a key determinant of magnesium excretion.34,35 In the DCT, transcellular reabsorption plays a key role. This is brought about by the TRPM6 and is influenced by calcineurin (by affecting NaCl cotransporter expression).35 Other channels that contribute to the electrochemical gradient in the DCT include the Na+/K+-ATPase voltage-gated chloride channels on the basolateral side and the voltage-gated potassium channels on the luminal side. These channels collaborate to promote potassium excretion into the lumen to maintain a relatively positive charge in the lumen, helping absorb magnesium (Figure 2). Notably, factors increasing the Na+/K+-ATPase or voltage-gated potassium channel activity such as the renin–aldosterone system can promote magnesium reabsorption.35,36 Being the orphan electrolyte of the body, magnesium reabsorption is modestly influenced by multiple hormones including insulin, parathyroid hormone, antidiuretic hormone, glucagon, renin, aldosterone, and calcitonin.1,24,37 Local prostaglandins, hypercalcaemia, and diuretics have also been noted to affect renal magnesium reabsorption in the tubules.37
HYPERMAGNESAEMIA
Epidemiology
Normal magnesium levels vary from 0.7–1.0 mmol/L or 1.7–2.4 mg/dL.31 Hypermagnesaemia is thus defined as magnesium levels equal to or greater than 1.1 mmol/L or 2.5 mg/dL. Levels greater than 2.0 mmol/L or 4.7mg/dL are considered critical. The incidence of hypermagnesaemia in hospitalised patients varies based on the study, from 3–5%38 and up to 10–12%.39,40 Incidence is higher in patients with suboptimal kidney function41 and in patients in the intensive care unit.40
Consequences of Hypermagnesaemia
Magnesium toxicity can range from an asymptomatic laboratory abnormality to widespread and sometimes fatal manifestations. The most common systems affected by magnesium toxicity are the nervous, cardiac, respiratory, and gastrointestinal systems. In mild cases, presentation might only involve nausea, light-headedness, and confusion. As magnesium levels rise above 7 mg/dL, the symptoms can become more serious with worsening drowsiness, decrease in deep tendon reflexes, blurring of vision from impaired pupillary accommodation, ileus, and bladder paralysis. It can also affect the cardiovascular system, causing hypotension and bradycardia. When magnesium levels rise higher than 12 mg/dL, life-threatening conditions ensue, including flaccid paralysis, respiratory depression, and cardiac arrhythmias, notably bradycardia and prolongation of the PR interval. This progresses to encompass fatal events with cardiorespiratory arrest or coma at magnesium levels ≥15 mg/dL.31
Hypermagnesaemia is associated with increased mortality. In a recent study of >14,000 patients, it was noted that the 30-day all-cause mortality was twice as high in patients with hypermagnesaemia compared to patients with normal serum magnesium levels.39
Causes of Hypermagnesaemia
The human body is well equipped to handle high loads of magnesium. Between bone magnesium buffering, controlled intestinal absorption, and, most importantly, the renal ability to precisely reabsorb only the required levels of magnesium, it is uncommon to find life-threatening hypermagnesaemia. To have hypermagnesaemia, intake must exceed excretory ability; thus, most cases of hypermagnesaemia occur in a situation of decreased kidney function with the inability, therefore, to excrete extra magnesium.42
Kidney disease
Kidney dysfunction, both acute and chronic, is possibly the most common cause of hypermagnesaemia. This is usually compounded by an increased magnesium intake. In one study, over 70% of patients with hypermagnesaemia had an underlying decrease in glomerular filtration rate (GFR).42 In a recent case series, 85% of patients with hypermagnesaemia had chronic kidney disease (CKD) and only 15% had normal creatinine levels.43 Factors that can decrease renal filtration of magnesium, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, non-steroidal anti-inflammatory drugs, adrenal insufficiency,24,31 hypothyroidism,24,31 and even hyperkalaemia (by causing bradycardia and hypotension in extreme cases), can cause worsening magnesium toxicity.20,35,44 Many cases of hypermagnesaemia occur in elderly patients with CKD on concomitant medications, like those mentioned above. Magnesium toxicity has been documented repeatedly in patients on lithium. The exact mechanism is unknown, but it is suspected to be driven by lithium-induced hypovolaemic state with a subsequent increase in renin and aldosterone stimulation leading to the increase in magnesium retention.24,31,45
Iatrogenic
The second most common cause of hypermagnesaemia is aggressive replacement or therapeutic usage of magnesium compounds. This is mostly seen in patients receiving intravenous magnesium infusions, such as obstetrics patients with pre-eclampsia or eclampsia.31 Hypermagnesaemia in these cases usually occurs despite normal kidney function since the rate of infusion exceeds the rate of renal excretion. Newborn infants of such patients have also been reported to have iatrogenic magnesium toxicity at birth, while some pre-term infants whose mothers were on magnesium-containing parenteral nutrition have also been reported to have symptomatic hypermagnesaemia. Magnesium-based bowel preparation regimens are some other commonly described iatrogenic causes of hypermagnesaemia.
Excessive absorption
Magnesium toxicity from increased intestinal absorption can occur either by surplus intake or prolonged intestinal exposure to the ingested magnesium content. Most cases of hypermagnesaemia, regardless of renal function, have exposure to exogenous magnesium in the form of magnesium-containing supplements, laxatives, antacids, cathartics, or enemas.44,46 Magnesium oxide, a commonly used laxative, is notorious for cases of toxicity in the elderly.43 The oral bioavailability of magnesium can vary from approximately 15% to 60%47 based on the ingested daily magnesium content. Magnesium oxide doses >1 g/day have been noted to increase risk of toxicity.48 Increased intestinal magnesium absorption can also be due to increased surface contact time between the magnesium and the intestinal cells, by a richer blood supply to the intestine, or decreased intestinal transit time. Thus, inflammatory bowel disease (through increased enteric blood supply) and constipation or ileus (through slower transit) can lead to hypermagnesaemia, regardless of the dietary magnesium content or normal kidney function. Both conditions lead to a vicious cycle, with increased magnesium levels slowing peristalsis and resulting in worsening ileus and increased intestinal absorption of magnesium.46,49 Medications causing constipation such as opiates and anticholinergics can precipitate hypermagnesaemia in the same manner.24 Vitamin D and its analogues cause increased magnesium absorption in the intestine and when coupled with CKD have resulted in cases of hypermagnesaemia.35,43
Rare causes
Magnesium toxicity has also been reported in cases of haemolysis, tumour lysis, rhabdomyolysis, and metabolic acidosis due to extracellular shifts of the intracellular magnesium stores. Sepsis, intestinal perforation, and intestinal ischaemia are some of the other uncommon conditions where hypermagnesaemia has been noted.35 Urethral irrigation with magnesium-rich hemiacidrin,24 which is used in nephrolithiasis management, has also been documented with cases of magnesium toxicity. Epsom salts (rich in magnesium) being used as laxatives have been reported as causes of hypermagnesaemia that require emergent hospitalisations.50,51 Calcium-alkali syndrome31 and familial hypocalciuric hypercalcaemia35,52 can result in magnesium toxicity because of decreased renal magnesium excretion. In familial hypocalciuric hypercalcaemia, there is an inactivating mutation in the calcium-sensing receptor gene (CaSR). This receptor is usually present in all segments of the kidney but most abundant on the basolateral side of the TAL. The CaSR usually regulates sodium chloride and divalent cation transportation both paracellularly and transcellularly; it does so by influencing multiple channels including NKCC2 and ROMK (normally inhibiting their activity in hypercalcaemic conditions) to help maintain a specific electrochemical gradient.53 An inactivating mutation of the CaSR causes hyperactivity of NKCC2 and ROMK and increases the positive charge in the lumen; this indirectly enhances paracellin activity, which eventually drives divalent cation transportation across the tubular cells and results in higher calcium and magnesium reabsorption both transcellularly and paracellularly.53 Calcium-alkali syndrome can also present as hypermagnesaemia caused by multiple indirect factors including magnesium-containing antacids, decreased intravascular volume, and acute kidney injury, all contributing to increased magnesium retention.54 Other rare causes include calcitriol (by increasing intestinal magnesium absorption)1 and ingestion of salt water55 from the Dead Sea, Western Asia (which has a high magnesium content). Conditions involving increased aldosterone, with or without increased renin levels, have also been linked with hypermagnesaemia. This happens due to aldosterone-mediated hyperactivity of Na+/K+-ATPase, ROMK, the sodium–chloride cotransporter, and voltage-gated potassium channels (in DCT), resulting in a higher positive charge in the tubular lumen and causing magnesium flow towards the basolateral side due to an increased electrochemical gradient.35,36 The causes of hypermagnesaemia are summarised in Table 1.
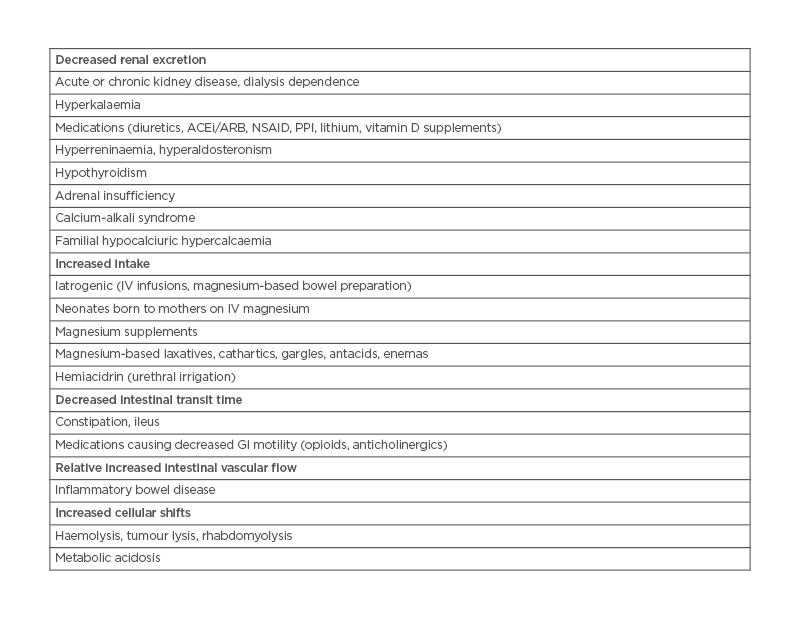
Table 1: Causes of hypermagnesaemia. ACEi: angiotensin-converting enzyme inhibitors; ARB: angiotensin receptor blockers; GI: gastrointestinal; IV: intravenous; NSAID: non-steroidal anti-inflammatory drugs; PPI: proton-pump inhibitors.
MANAGEMENT
Most of the cases of hypermagnesaemia are asymptomatic to mildly symptomatic, yet an increased mortality rate has been noted in patients with hypermagnesaemia who are admitted to the hospital. Identifying and removing the culprit agent is the foremost step to the management of hypermagnesaemia. Asymptomatic cases with a normal GFR do not require specific treatment, except from withholding all magnesium-containing medications; thereby, excess magnesium is usually excreted in the urine. The half-life of magnesium in the body with a functioning kidney is approximately 28 hours.
In settings of acute symptomatic toxicity, it is essential to rapidly lower magnesium levels. Medical management is usually also required in patients with hypermagnesaemia with CKD and estimated GFR (eGFR) between 15 and 45 mL/min/1.73m2. The foremost step is to administer a dose of 100–200 mg of elemental calcium over 5–10 min to rapidly counteract the neuromuscular and cardiovascular ramifications of hypermagnesaemia. This can be achieved by administering either calcium chloride or calcium gluconate intravenously. Magnesium renal excretion can be augmented with the help of loop diuretics, such as furosemide at a dose of 1 mg/kg. This dose can be increased depending on eGFR. Magnesium excretion with diuretics is further aided by adding intravenous isotonic fluids, such as saline at 150 mL/hour.
Dialysis is indicated in cases of severe (levels >6 mg/dL), symptomatic hypermagnesaemia, where patients have critical symptoms or anuria. Patients with an eGFR usually <15 mL/min/1.73m2 and hypermagnesaemia also often require treatment with dialysis. In such cases, medical management should be started immediately while dialysis is being established. Haemodialysis is preferred over peritoneal dialysis because it reduces magnesium levels faster; haemodialysis can be expected to remove 50% of the magnesium load in a 4-hour session.31
CONCLUSION
Magnesium is one of the most important electrolytes in the body. While justified emphasis is given to hypomagnesaemia and its role in cardiac arrhythmias, hypermagnesaemia can have equally debilitating effects. Hypermagnesaemia can have widespread effects in the body and affects the nervous, cardiovascular, respiratory, and gastrointestinal systems. It can be a cause of altered mentation, ranging from somnolence to coma, and other fatal conditions, ranging from respiratory failure to cardiac arrhythmias. Often, magnesium toxicity is the result of the co-administration of multiple medications, which individually might be safe but when given together, especially in patients with predisposing conditions like kidney disease, can result in adverse events. It is not only prudent to identify the signs and symptoms of magnesium toxicity early and reverse the disorder, but equally valuable to be familiar with the causes of hypermagnesaemia. Distinguishing the magnesium-rich medications or those with the potential to cause an increase in magnesium absorption or decrease in magnesium excretion must be a part of daily medical practice; this is imperative to prevent harm occurring to the predisposed patient population at high risk of developing magnesium toxicity.