Interviewees: Mårten Segelmark,1 YKO (Onno) Teng2
1. Department of Nephrology, Lund University, Lund, Sweden
2. Department of Nephrology, Leiden University Medical Center, Leiden, the Netherlands
Disclosure: Prof Segelmark has received consultancy fees from ChemoCentryx and research grants from Hansa Biopharma. Dr Teng has declared no conflicts of interest.
Support: The publication of this interview feature was supported and reviewed by Vifor Pharma.
Acknowledgements: Medical writing assistance was provided by Dr Juliet George, Chester, UK
Citation: EMJ Nephrol. 2020;8[Suppl 2]:2-10.
Interview Summary
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) comprises a group of rare, necrotising inflammatory diseases. AAV can cause damage to blood vessels throughout the body, including in the kidneys, lungs, and intestines, and is typically treated with immunosuppressive and immunomodulatory therapies to help induce remission. However, the pathophysiology underlying this multifactorial condition is not completely defined. During interviews conducted by EMJ in early 2020, two experts in the field of nephrology and autoimmune disease, Prof Mårten Segelmark and Dr Onno Teng, discussed the current understanding of the pathological processes involved in the damage associated with AAV. This included key interactions in the immune pathways that lead from the initial stage of ANCA production to the resulting vessel inflammation. In particular, the experts highlighted the critical but often overlooked role of complement and neutrophils in this process. The importance of this knowledge for improving the treatment and care of patients with AAV was also emphasised.
THE MECHANISMS OF DAMAGE IN ANTINEUTROPHIL CYTOPLASMIC ANTIBODY-ASSOCIATED VASCULITIS
AAV is characterised by distinct pathologic lesions linked to the production of ANCA. With classification based on pathological as well as clinical features, the main forms of AAV include microscopic polyangiitis, granulomatosis with polyangiitis ([GPA] previously known as Wegener’s granulomatosis), and eosinophilic GPA (previously known as Churg–Strauss syndrome).1 The damage and inflammation in AAV predominantly affects small blood vessels, with localised necrosis of vessel walls observed across multiple body systems. Because of this, glomerulonephritis, inflammation of the glomerular capillaries of the kidneys, is another common component of the AAV diseases. Whenever disease pathology is limited to glomerulonephritis alone, with no evidence of systemic inflammation, physicians classify the disease as renal-limited vasculitis.1,2 Together, the effects of AAV can result in systemic organ damage and failure, with the kidneys and lungs as major targets.3
Underlying these effects, AAV has a complex pathophysiology that is generally divided into two stages: ANCA development (i.e., antibody response) and damage processes. In Stage 1 of AAV, the body loses its tolerance to ANCA antigens and B cells produce ANCA in an autoimmune response. ANCA is most commonly directed against the neutrophil lysosomal enzyme proteinase 3 ([PR3] in GPA) and myeloperoxidase ([MPO] in microscopic polyangiitis),1 but other targets have been reported, such as lysosome-associated membrane protein. Stage 2 comprises the active vasculitic process and associated damage or inflammation, which appears to be driven by the priming of neutrophils and their activation and interaction with the complement system.4,5
Focussing on the roles of neutrophils and the complement pathway in the route to vasculitic damage in AAV, the experts agreed that knowledge was incomplete. “It is well known that both neutrophils and complement are very important for the disease process at different levels, but their involvement is not completely understood,” said Prof Segelmark. “Ultimately, we know that antibodies are the end product of an autoimmune response, where some type of autoantigen has triggered the immune system, leading to B-cell responses that prompt autoantibody production. While we can measure those autoantibodies, the associated processes are less defined,” added Dr Teng.
THE ROLE OF NEUTROPHILS
Discussing evidence for the involvement of neutrophils in initiating the disease process, Dr Teng explained: “We know that AAV is very much related to neutrophils because the antigens that cause the ANCA autoantibodies (PR3 and MPO) are both enzymes that you find in the cytoplasmic granules of neutrophils. People ask why AAV patients make antibodies against these enzymes, some of which are enclosed in a granule, inside a neutrophil. How are they exposed to the immune system? The standard way of thinking of this is that there is an unknown neutrophil trigger factor, ‘X’, that leads to massive release of the enzyme-containing granules. PR3 and MPO would then be exposed outside the cell. This excessive quantity would mean the enzymes couldn’t be cleared quickly enough so, at some point, they would be picked up by an immune cell as an autoantigen. However, as most of the PR3 and MPO is displayed on the plasma membranes of neutrophils, it’s still a very difficult concept to understand how these antigens are presented to the immune system.” Dr Teng continued: “About 15 years ago, it was discovered that neutrophils can produce neutrophil extracellular traps (NET), which are extracellular strands of DNA.6 If you analyse this extracellular DNA you find that all the enzymes that are normally in the cytoplasm of neutrophils, including MPO and PR3, are actually on the DNA of these NET. So now you have a highly inflammatory situation in which the immune system is triggered by extracellular DNA where it will also pick up all the MPO and PR3 proteins displayed on these NET. By harbouring the autoantigens, NET trigger a humoral autoimmune response, leading to ANCA development.”
Dr Teng went on to explain that this process of ‘NETosis’ is a physiological mechanism that occurs in every healthy person when neutrophils encounter bacteria, viruses, or fungi. “It is a very quick reaction, a sort of kamikaze response, in which the neutrophil throws out its DNA,” he stated. “Because neutrophils are most abundantly present in the circulation, it seems plausible that for this encounter to take place, the bacteria must be within the circulation, i.e., a very severe infection. When a person has a severe infection with pathogens in the bloodstream, you want your immune system (and, in this case, your neutrophils) to elicit a fast response. NETosis is one of the neutrophil’s quickest responses.7 It extrudes its own DNA strands, which can trap bacteria and contain the infection.” Crucially, in AAV, when there are no bacteria to elicit this response, NET still appear to be activated. According to Dr Teng: “If serum from AAV patients is added to healthy neutrophils, they will form an excessive amount of NET. We don’t understand the exact trigger but we know that it’s not ANCA because the excessive NET formation is still induced when ANCA is taken out of the serum. Moreover, we’ve shown that when you take serum from a lot of AAV patients and mix this with neutrophils, these neutrophils become massively activated, more so than if you use serum from healthy controls. There is also an association with the level of disease activity, as the more active the patient’s disease, the more NET that are induced,” he said. Furthermore, Prof Segelmark pointed out that it has been shown that neutrophils from patients with AAV, including those without signs of ongoing inflammation, are intrinsically prone to undergo NETosis, both spontaneously8 and when stimulated with ANCA.9 This is mirrored by a decreased tendency to undergo spontaneous apoptosis, a mode of cell death that is not associated with tissue damage.10
Prof Segelmark and Dr Teng then considered the current evidence for the involvement of neutrophils in the vasculitic damage observed in patients with AAV. “It is known that neutrophils are extremely critical in all forms of small vessel vasculitis; eosinophils may also be involved but neutrophils are the key culprits in causing the damage, at least that’s my opinion,” began Prof Segelmark. He cited early studies using animal models of glomerulonephritis that identified neutrophils as a necessary cellular component of glomerular injury,11 and added: “In the glomerulonephritis in AAV, immune complexes are present but it’s the neutrophils chewing up the basement membrane, leading to bleeding, and entering the extravascular space that triggers the process of their formation.”
The experts then considered how the different neutrophil-related mechanisms, including NETosis, may contribute to the vessel damage in AAV. “Of course, without adhesion there wouldn’t be any neutrophils there, so that is very important. Then the neutrophils need to be activated and release their contents by exocytosis or by NETosis,” said Prof Segelmark. “Activated neutrophils will express all sorts of proteins that make them adhere to the vessel wall,” continued Dr Teng, who also cited the generation of reactive oxygen species, including those derived from MPO, as being a key mechanism in triggering NET release. “We know that after a few hours of activation these neutrophils will explode and extrude their NET. Once neutrophils go into NETosis they start to adhere more to the vessel wall, as do a lot of immune cells, eventually causing the small-vessel destruction.” Prof Segelmark concluded: “I cannot tell you what would happen if NETosis could be blocked completely, whether that would prevent the disease manifestations of AAV, but it seems logical that NETosis is important.”12
THE ROLE OF COMPLEMENT
It has been suggested that the combination of complement and neutrophils is a pivotal but under-recognised partnership in the pathophysiology of AAV, and Prof Segelmark gave an overview of evidence for the involvement of complement in AAV disease processes. “The first good models of AAV used rodent knockouts to identify the various parts of the complement system that were necessary for disease.13,14 It became apparent that it was not the terminal pathway (to vascular damage) that was the most important element, but rather the recruitment of the neutrophils by the complement system that was essential,” he explained. “In addition, there is circumstantial evidence. In the 1990s, before the genomic era, we found that there were genetic variants in the effect of C3 component: C3F and C3S. The C3F allele was shown to be more prone to activation than C3S, which is the more common allele, and there was a higher-than-expected frequency of the C3F allele in patients with systemic vasculitis.15,16 Later, these alleles were also shown to be influential in the outcome of renal transplantation17,18 and the occurrence of age-related macular degeneration.”19,20 Components C3 and C5 are common to all three pathways that activate the complement system: the classical, lectin, and alternative pathways (Figure 1). The experts agreed that it is the self-sustaining alternative complement system that seems to be chiefly involved in the pathophysiology of AAV. Under nonactivated conditions, the alternative pathway is maintained at a low level of C3 autoactivation, known as ‘tick-over’, by control proteins. Once triggered, this autoactivation loop is amplified, and C3 is converted to C3b and C3a by a different factor from that used by the classical and lectin pathways. In turn, C5 is activated to form the potent chemoattractant C5a, as well as the cytotoxic membrane attack complex C5b-9. The result is amplification of the inflammation response that continues until it is specifically downregulated by other complement factors. Dr Teng explained: “C5 is the common effector pathway of the complement system and C5a, a by-product of C5 activation, is a highly proinflammatory molecule. Preclinical models showed that if you block C5, there is a lot of disease improvement.”13
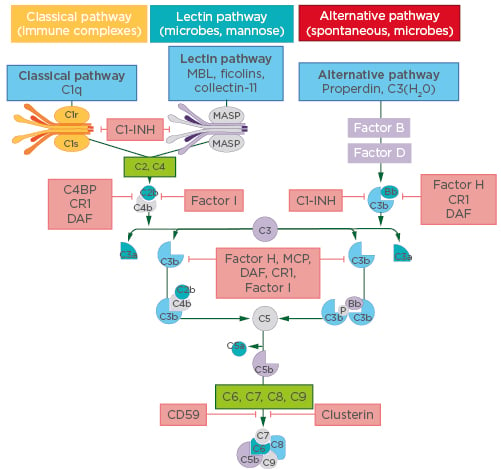
Figure 1: The three complement pathways.
C1-INH: C1 inhibitor; CR1: complement receptor 1; C4BP: C4b-binding protein; DAF: decay-accelerating factor; MASP: mannose-binding lectin-associated serine protease; MBL: mannose-binding lectin; MCP: membrane cofactor protein.
Adapted from Poppelaars F et al., 2018.21
Reflecting on potential differences in the regulation of this process in AAV, Prof Segelmark commented: “Our current knowledge doesn’t point to anything being different in the mechanisms of the alternative pathway in AAV; rather, it might be that they are more active, i.e., appropriate activation of the alternative pathway by dying neutrophils or vascular endothelial damage can still be detrimental if not controlled.” Dr Teng expanded on this point: “One of the most important molecules that is needed for excessive complement pathway activation is a protein called properdin. For the alternative pathway to be activated, C3 must be bound to a cell membrane and this binding is mediated by properdin. The interesting part about properdin is that it is, itself, a molecule of the complement system that is produced by myeloid-derived cells, macrophages, or neutrophils, which are the primary responders for inflammation. So, in inflamed tissues, including in AAV, there are a lot of macrophages and neutrophils to produce properdin and ensure that C3 is bound to membranes locally in the tissue, and thus activate the alternative complement system locally.” This local activation aspect of the complement response is characteristic of AAV, and one which is highly interconnected. “Local activation of the alternative pathway by the complement system brings about C5a release. The main task of C5a is to induce chemotaxis of the macrophages and neutrophils to the inflamed area which, in turn, produce a lot of properdin causing C3 activation, and then again, more C5a. It is a vicious circle,” concluded Dr Teng.
COMPLEMENT IN TREATMENT AND DIAGNOSIS
Given the clear evidence for involvement of complement, and its potential overactivation in AAV, a frequent question is how this fits with the glomerulonephritis in AAV appearing to be pauci-immune, with little evidence of complement on biopsy. “First of all, pauci-immune does not mean that there is nothing,” explained Prof Segelmark. “It means that it is not abundant, or the prominent element. Here we have a process where the complement system seems to be activated on the surface of neutrophils, but neutrophils are not present at inflammatory loci for more than maybe 24 or 48 hours. So, this may be a transient activation that does not lead to a long-standing deposition, unlike immune complexes which seem to get stuck in the glomeruli,” he clarified.
Adding to this reasoning, Dr Teng stated: “The fact that there are not many antibodies seen in the kidney tissue at biopsy in AAV, even though kidney inflammation is one of the hallmarks for diagnosing the disease, again points to involvement of the alternative, as opposed to the classical, complement pathway. Studies have indeed confirmed that C3 deposition in kidney biopsies of AAV patients associates with worse prognosis.22 However, then we have to ask how the alternative complement pathway is activated? Here, again, I think it’s more a case of local tissue complement activation than systemic activation.”
Both experts noted how this predominance of complement in the tissues presents a challenge in terms of measuring its levels during patient diagnosis or treatment in AAV. Prof Segelmark explained: “The normal way that complement is measured is testing for the level of lytic activity in the serum. To make it more meaningful (in AAV) we need other tools, other measures of complement activation that could be more useful and here, I think, the jury is still out. There is a lot of literature on complement products and breakdown products in AAV,23,24 but, as yet, we do not know how to use this information clinically. Standard assays that have been developed for systemic lupus erythematosus (SLE), for example [which measure complement levels in the serum] are not really useful in AAV.” Dr Teng agreed with this view. “In most patients, you don’t see complement systemically, so, for a clinician, this measurement is not relevant. For a research scientist it could be interesting to assess the minority of patients who do show systemic activation of the alternative pathway. However, even then, measuring systemic complement and activation is less informative than measuring complement biology at the tissue level,” he said.
OTHER CELL INVOLVEMENT
Concerning the involvement of other cells in the pathophysiology of AAV, Prof Segelmark commented: “It is likely that other cells come in later, after the ‘early-ish’ initiation timepoint when neutrophils or eosinophils are, I think, the key players. After breakdown of the integrity of the vascular wall, there will be bleeding and there will be coagulation. As yet, we do not really know the role of thrombocytes; they are present, they are activated under these circumstances, and they have a lot of substances to release, so coagulation might play a part. However, at this early stage, my view is that T cells or B cells are not that important locally,” he said. Dr Teng agreed that the involvement of other cells comes later in the process, “Many different cell types are attracted to the damaged vascular wall. You get chemotaxis of monocytes and macrophages, local activation of complement, more chemoattractants, and then you get a secondary immune response with T cells and eventually B cells going into the area of vascular damage. It is the start of a whole self-fulfilling mechanism,” he said.
THERAPEUTIC TARGETS
Having considered underlying evidence for the complement–neutrophil interaction in AAV, the experts moved on to discuss whether this represents a relevant investigational target in AAV. “Both theoretically and also from the animal models and clinical data that have been generated, it seems logical to target the neutrophils in some way,” stated Prof Segelmark. “There are probably lots of other ways to do this but targeting the complement–neutrophil interaction is certainly one of them,” he suggested.
Dr Teng also supported the therapeutic relevance of the complement–neutrophil interaction. “A year ago, we would have queried whether complement inhibition would be beneficial for AAV patients,” he began, “but now we have Phase II and Phase III studies showing the activity of a C5a-inhibiting compound in patients with AAV: the CLEAR and ADVOCATE studies.25,26 So now we can say that there is definitely complement activation in AAV, and targeting complement, at least, complement proteins, is something that is useful for patients. However, do we know exactly how this complement inhibition improves the disease mechanism? This is still an open question,” said Dr Teng. Continuing this point, Prof Segelmark commented: “We don’t even know, completely, how current therapies like cyclophosphamide [an immunosuppressant] or rituximab [a monoclonal antibody to CD20] work in AAV. So, there are a lot of unknowns. Complement inhibition would seem to stop the vasculitic process and prevent new damage occurring. Of course, there are other possible processes coming in later, after the first 24 hours, that could be important to target in order to get the right kind of healing to occur, to prevent fibrosis, and prevent the accumulation of debris. However, I think that if we could prevent the interaction between the neutrophils and complement, it is likely that the neutrophils would be less numerous at the site, that they would be less activated, and that they would produce much less damage,” he proposed.
Dr Teng expanded on this by explaining, “Complement is part of the acute inflammatory reaction we see in AAV and C5a is a mediator of a lot of inflammation. As anti-inflammatory agents, steroids have been the backbone of our treatment for AAV for decades. At this point, I think that C5a inhibition could be as effective as steroids in reducing that inflammatory reaction, as the Phase II CLEAR study showed that a complement (C5a) inhibitor could replace steroid use.25 Although we haven’t seen full Phase III data yet, findings appear positive versus standard steroid treatment.26 For the patient and the clinician, I think it would be a major breakthrough if you could treat AAV patients without using steroids, especially at the beginning when the inflammation in AAV is really severe. We tend to use very high doses of steroids at this point, when there is risk of pulmonary haemorrhage and kidney failure, so there is less concern about the steroid side effects.” According to Dr Teng, having an alternative would mean being able to consider the side effects of a treatment even when selecting therapy for patients at the most high-risk stages of disease. “This would obviously be a benefit for patients, and also for clinicians who have to keep these patients on steroids and manage compliance, despite all the side effects,” he said. While Prof Segelmark shared this view, he also cautioned about the consequences of completely blocking the complement pathway. “The complement system is very important in many respects, removing debris and preventing infections, for instance. Every time you ‘tune down’ a system, there are side effects. In the acute situation you may be afraid of some type of opportunistic infection; if there is a long-term break in the system then impeding the clearance of dead cells and debris in the body may cause issues, and could perhaps trigger SLE-like symptoms. There is always a danger when you start altering important systems in the body, but I think that halting the vasculitic process in this way could be a very promising target,” he said.
Dr Teng agreed that while having great potential, complement inhibition may not be the complete solution, giving his reasoning from a different angle. “From the clinical point of view, of course the benefit is that you have a more targeted treatment. From a scientific point of view, the counterargument is also true. We are used to general immunosuppression where we just suppress the whole immune system; now we have a targeted approach which might be very potent, but it may not suppress the immune system well enough. For instance, it could be that once the inflammation is effectively reduced, another aspect of the immune system or another mechanism that causes inflammation is not targeted. So, I think you have to ask whether this is the solution for everything in AAV, or if some other part of the immune system is being missed.”
Dr Segelmark added: “There are many development studies in other [non-AAV] forms of glomerulonephritis that are focussing on different parts of the complement system. There is a lot of ongoing research, and there are definitely aspects that would be of interest to evaluate also in relation to AAV.” “Complement inhibition is progress, but it also raises a lot of questions,” concluded Dr Teng.
CONCLUSION
AAV has an intricate pathophysiology that, consistent with its name, may be perceived as predominantly related to the activities of ANCA autoantibodies. However, as Prof Segelmark and Dr Teng described, the combination of the alternative complement system and neutrophils is integral to disease development, from the triggering of ANCA production through to generation of vasculitic damage (Figure 2). Although these interactions are not yet clearly defined, both preclinical and clinical data support their involvement in the inflammation and damage that is characteristic of AAV. Moreover, such studies have revealed evidence of highly interrelated pathways that can be blocked at key points to alleviate disease processes. The experts agreed that while the actions of complement and neutrophils in AAV are not yet fully understood, targeting this intricate partnership could help deliver new, potentially better-tolerated, approaches to treatment.

Figure 2: Key stages in antineutrophil cytoplasmic antibody-associated vasculitis and the role of complement and neutrophils.
ANCA: Antineutrophil cytoplasmic antibody; MPO: myeloperoxidase; NET: neutrophil extracellular traps; PR3: proteinase 3.
Biographies
Prof Mårten Segelmark
Lund University, Lund, Sweden
Prof Giannini is an expert in liver pathophysiology with application to the prognostic study of the liver functional reserve. His research interests include the noninvasive staging of chronic liver disease, haemostasis in liver patients, staging, and therapeutic management of hepatocellular carcinoma. He is author of >250 publications in international journals and has written several book chapters.
Prof Mårten Segelmark is a specialist in internal medicine and nephrology who trained at Lund University Hospital in Lund, Sweden, and completed postdoctoral training at the University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. He is now Professor of Nephrology at the Linköping, Sweden, and Lund Universities. He served as President of the XIVth International ANCA and Vasculitis Workshop in 2009, and as President of the Swedish Society of Nephrology (SNF) 2012–2013. Since 2016, he has been the Vice President of the Immunonephrology Working Group of the European Renal Association–European Dialysis and Transplant Association (ERA–EDTA). Prof Segelmark has published 111 original articles and 37 reviews and book chapters.
Dr YKO (Onno) Teng
Leiden University Medical Center, Leiden, the Netherlands
Dr YKO (Onno) Teng is a nephrology clinician-scientist at the Department of Internal Medicine of the Leiden University Medical Center (LUMC) in Leiden, the Netherlands. He is head of the nephrology outpatient clinic and coordinator of the Leiden outpatient clinic for Lupus, Vasculitis, and Complement-mediated Systemic diseases (LuVaCs) which accommodates top referral, multidisciplinary, tertiary care on a regional and national level. He also leads and coordinates a clinical, multidisciplinary pathway aimed at counselling and guiding a pregnancy wish for patients with SLE or antiphospholipid syndrome.