Abstract
Sodium–glucose co-transporter inhibitors (SGLT2i) have recently gained a lot of emphasis in their role in preventing progression of chronic kidney disease and helping with cardiac mortality. Various studies have proven the benefit of these medications in the management of patients with kidney and heart disease. SGLT2i exert their effect in the proximal convoluted tubule with various downstream effects noted in the kidney also. With spreading use of these medications, it is imperative to understand the effects they have on various electrolytes and the pathways involved in bringing about these changes in the kidney. Here, the authors review the current knowledge of SGLT2i with their effects on the kidney, electrolytes, and water balance.
Key Points
1. Sodium–glucose cotransporter inhibitors (SGLT2i) exert their effect in the proximal convoluted tubule, with various downstream effects, with benefits extending beyond glycosuric effects to a role in preventing progression of chronic kidney disease and help with cardiac mortality.2. The renal benefit of SGLT2i in preventing chronic kidney disease progression likely occurs via several mechanisms, including decreased glomerular hypertension by blocking sodium reabsorption in the proximal convoluted tubule and decreased glucose resorption in the nephron.
3. The renal effects of SGLT2i can affect biochemical and water balance, including causing an overall free water loss alongside decreased uric acid and increased magnesium levels.
INTRODUCTION
Sodium–glucose co-transporters (SGLT) play an important role in glucose transport across epithelial cells. SGLT1 plays a major role in intestinal glucose absorption, while SGLT2 reabsorbs the majority of the filtered glucose in the proximal convoluted tubule (PCT).1 In 2011, blockage of SGLT2 in the PCT showed an increase in urine glucose in healthy subjects.2 One year later, canagliflozin was shown to improve glycaemic control in patients with Type 2 diabetes (T2D).3
Over the past few years, there have been numerous landmark trials supporting the utility of SGLT2 inhibitors for patients with heart failure and chronic kidney disease (CKD).4-6 From the kidney perspective, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines have been adjusted to include SGLT2 inhibitors as first line therapy along with metformin for patients with diabetic kidney disease.7 Dapagliflozin was also recently approved by the U.S. Food and Drug Administration (FDA) for use in high progression risk CKD patients, regardless of diabetes status. As the utility of SGLT2 inhibitors becomes more prevalent, we continue learning more about their effects on the kidney, heart, electrolytes, and other biochemicals. This review will cover the current knowledge on the renal effect, and the pathophysiological mechanism of this novel class of medications.
CHRONIC KIDNEY DISEASE
The earliest of the landmark trials that showed a potential utility of SGLT2 inhibitors (SGLT2i) in CKD was the EMPA-REG trial in 2015.4 This trial showed that treatment with SGLT2i provides kidney protection for patients with CKD. Equally far-reaching trials followed over the next few years, including CREDENCE5 and DAPA-CKD.6 CREDENCE demonstrated the benefit SGLT2i provide in patients with diabetic kidney disease, while DAPA-CKD showed their utility in non-diabetic proteinuric CKD. It is now well established that the beneficial effects of SGLT2i go far beyond their glucosuric effects.
Glomerular hypertension is one of the maladaptive mechanisms that can lead to progressive CKD. The impact of SGLT2i on glomerular hypertension is now an accepted mechanism that could underscore their kidney protective effects. By blocking sodium reabsorption through SGLT2 in the PCT, there is a natriuretic effect that induces tubulo-glomerular feedback, resulting in the vasoconstriction of the glomerular afferent arteriole. This leads to decreased glomerular hyperfiltration and decreased glomerular hypertension.8
In addition to their effect on reducing glomerular hypertension, SGLT2i also decrease the amount of glucose being reabsorbed in the nephron. This is hypothesised to exert a protective effect against matrix expansion, interstitial fibrosis, and macrophage infiltration,9 all of which are thought to be by-products of glucose-induced oxidative stress.10,11 SGLT2i are also believed to provide kidney protection through generating a systemic low ketone state. In what has become known as the ‘Thrifty Substrate Hypothesis’, SGLT2 inhibitors generate a state of mild, persistent hyperketonaemia. Ketones are then oxidised, preferentially over free fatty acids in organs such as the heart and kidney, thereby generating less reactive oxidative species and free oxygen radicals when compared to the consumption of free fatty acids.12,13
SODIUM
While seemingly simple, the impact of SGLT2i on sodium balance is more complicated than the increased excretion resulting from blockage of the SGLT2 receptors.
SGLT2 is a co-transporter that transports one sodium ion for each molecule of glucose. In contrast, SGLT1 transports two sodium ions per molecule of glucose.14,15 The location of these SGLT transporters in the PCT plays a role in the net impact of SGLT2i on sodium balance. Since SGLT1 transporter is located distal in the PCT (in the S3 segment for SGLT1 versus in the S1 and S2 segments for SGLT2), the action of SGLT2i increases the delivery of glucose to the S3 segment of the PCT. This results in an increase in SGLT1 action. From a sodium standpoint, blocking the SGLT2 transporter will result in decreased SGLT2 activity which reabsorbs one sodium molecule per glucose. However, by doing so, more glucose is available to be reabsorbed through the SGLT1 receptor, with resultant two sodium molecules reabsorbed per glucose.16
Other than SGLT 1 and 2, there has been evidence that SGLT2i cause the inhibition of sodium–hydrogen exchanger 3 (NHE3), which exchanges one sodium ion per hydrogen molecule. SGLT2i have been shown to directly inhibit NHE in cardiac cells in animal studies.17,18 In the renal tubular cells, SGLT2 interacts with NHE3 through the action of membrane associated protein 17,16 which results in decreased sodium reabsorption in the PCT. Overall, there seems to be a net negative sodium balance mediated through the inhibition of both the SGLT2 transporter, as well as the decreased action of NHE3 (Figure 1).
Figure 1: Summary of sodium–glucose co-transporter 2 inhibitor effects on sodium balance.
ENaC: epithelial sodium channel; Glu: glucose; GLUT2: glucose transporter 2; K: potassium; MAP17: membrane associated protein 17; Na: sodium; NHE3: sodium hydrogen exchanger 3; RAAS: renin–angiotensin–aldosterone system; ROMK: renal outer medullary potassium channel; SGLT: sodium–glucose co-transporter; SGLT2i: sodium–glucose co-transporter 2 inhibitor.
Clinically, SGLT2i have shown a measurable yet transient increase in patients’ urine output and urinary sodium excretion.19 Measurement of plasma volume to determine the overall sodium balance that occurs as a result of SGLT2i therapy showed conflicting results due to the variation in the methods of measurement. For example, when bioimpedance spectroscopy was used, there was no measurable change in total volume.20 However, when total volume was measured using indicator dilution with I-131 albumin, a clear drop in total plasma volume was observed.21
Despite these mixed findings, a clear and well-established result of SLGT2i is a reduction in both systolic and diastolic blood pressures.22-24 This change in blood pressure, albeit only mild, points towards an overall net negative sodium balance, rendering these agents effectively as weak diuretics.
MAGNESIUM
Hypomagnesaemia is an electrolyte imbalance that is observed in a large segment of patients who suffer from T2D.25 This hypomagnesaemia has been associated with a more rapid decline of renal function,26 higher rates of cardiovascular disease,27,28 and is even a predictor of end-stage renal disease in these patients.29 SGLT2i have been shown to increase serum magnesium in patients with T2D,30 but the pathophysiology is still poorly understood.
Several mechanisms have been proposed to explain this increase in serum magnesium upon SGLT2i treatment, including haemoconcentration, resulting from the diuretic effect of SGLT2i; an increase in glucagon levels, which results in increased magnesium absorption in the distal convoluted tubules; a decrease in insulin levels, resulting in a decreased shift of magnesium intracellularly; and an increase in insulin sensitivity, which can result in an increased expression of the TRPM6 channel that further decreases magnesium urinary excretion.25,31
URIC ACID
Patients with T2D tend to have elevated uric acid levels which are associated with an increased risk of cardiovascular disease32 and kidney disease. Hyperuricaemia has also been linked to faster CKD progression,33-35 although urate-lowering therapy has not been shown to provide kidney benefits.36,37
SGLT2i seem to carry a urate-lowering effect beyond what would be expected as a result of the osmotic drive.38 There is evidence that suggests that SGLT2 inhibition results in increased tubular secretion of uric acid that is mediated through glucose transporter (GLUT9) isoform 2 and urate transporter 1 (URAT1).39 Increased urinary glucose availability with SGLT2 blockade leads to activating GLUT9 isoform 2 for glucose reabsorption as opposed to urate reabsorption, ultimately leading to a uricosuric effect. It has also been hypothesised that GLUT9 isoform 2 activation may also facilitate urate secretion.40,41 The role of URAT1 is not well understood, but it is postulated to reabsorb urate under physiological conditions. SGLT2i downregulate URAT1, leading to more uricosuria (Figure 2).39
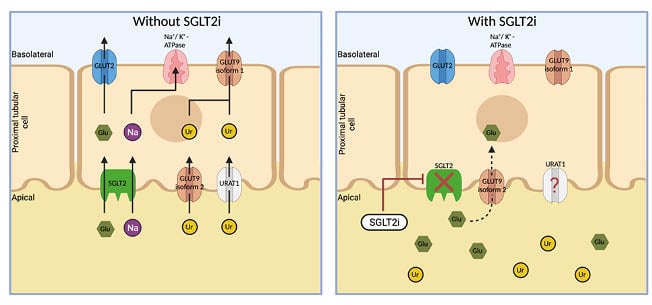
Figure 2: Visual representation of sodium–glucose co-transporters 2 inhibitor effects on urate.
The increased availability of glucose causes GLUT9 isoform 2 to reabsorb glucose rather than urate. URAT1 seems to be downregulated when SGLT2i are used; however, the mechanism behind this is not entirely clear. Glu: glucose; GLUT9: glucose transporter 9; K: potassium; Na: sodium; SGLTi: sodium–glucose co-transporter; SGLTi: sodium–glucose co-transporter inhibitor; Ur: urate; URAT1: urate transporter 1.
When looking at large patient analyses, there has been a considerable decrease in uric acid levels in patients receiving a SGLT2i,42 as well as a potential protection against the incidence of gout in these patient populations.43
PHOSPHOROUS AND CALCIUM
Phosphate levels, fibroblast growth factor 23, and parathyroid hormone have been shown to increase in patients who are on SGLT2i.44,45 It is speculated that the increased phosphate levels are related to an increase in reabsorption through sodium phosphorous co-transporters IIa and IIc in the PCT, due to the increased abundance of sodium ions in the urinary space upon SGLT2 inhibition (Figure 3).41
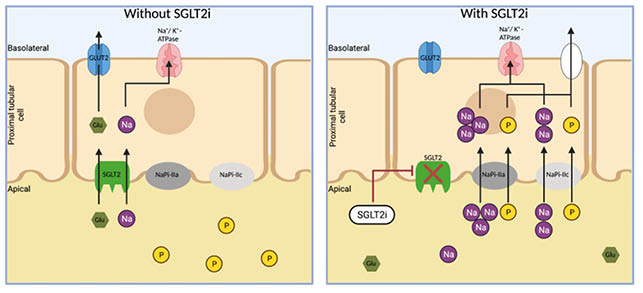
Figure 3: Visual representation of sodium–glucose co-transporter 2 inhibitors effects on phosphorous.
The increased availability of sodium will cause increased activation through NaPi IIa and IIc. Glu: glucose; GLUT2: glucose transporter 2; Na: sodium; NaPi: sodium phosphorous co-transporters; P: phosphorous; SGLT: sodium–glucose co-transporter; SGLTi: sodium–glucose co-transporter inhibitor.
Although there is evidence of an increase in fibroblast growth factor 23, parathyroid hormone and urinary calcium levels44 with the use of SGLT2i, the overall effect on serum calcium levels appears minimal.45
WATER
SGLT2i have an interesting effect on water balance, as they cause an increase in plasma sodium.46 This overall free water loss is believed to occur as a result of osmotic diuresis caused by the urinary loss of glucose and sodium.47 This class of medications has shown a potential utility in increasing free water loss, and resulting in an increased serum sodium in patients with syndrome of inappropriate antidiuretic hormone secretion.46 There are also ongoing investigations to determine if the SGLT2i empagliflozin can provide benefit in patients with euvolemic and hypervolemic hyponatraemia.
POTASSIUM
The effect that SGLT2i have on potassium balance is not very clear. There is some evidence that canagliflozin in particular can result in changes in potassium, but other SGLT2i do not exhibit the same effect.48 It seems as though canagliflozin can decrease the risk of hyperkalaemia when compared with placebo in most conditions.49 However, there have been reports of hyperkalaemia as well in specific conditions, where higher doses of canagliflozin are given in addition to renin–angiotensin–aldosterone system blockade and/or a potassium-sparing diuretic in patients with a reduced estimated glomerular filtration rate.50 It is possible that the initial drop in estimated glomerular filtration rate could lead to hyperkalaemia in the setting of advanced CKD; however, in the long-run, there seems to be a signal towards less hyperkalaemia.
CONCLUSION
In short, SGLT2i have revolutionised the management algorithms of CKD and heart failure. They have clear protective kidney effects in proteinuric, and possibly non-proteinuric, CKD, regardless of diabetes status or its control. Understanding of how these agents exert their protective effect is growing steadily and healthcare professionals are learning more about how they affect water and the various electrolytes. While this understanding has grown immensely in recent years, there are many questions and gaps in understanding that remain to be addressed.
SGLT2i seem to have an overall natriuretic effect, as well as an overall free water loss. They cause uric acid levels to decrease while causing magnesium levels to increase. Given the continued expansion in the use of SGLTi, it is becoming increasingly important for prescribers to realise how these revolutionary agents can contribute to different multiple facets of the patients’ metabolic profiles (Table 1).
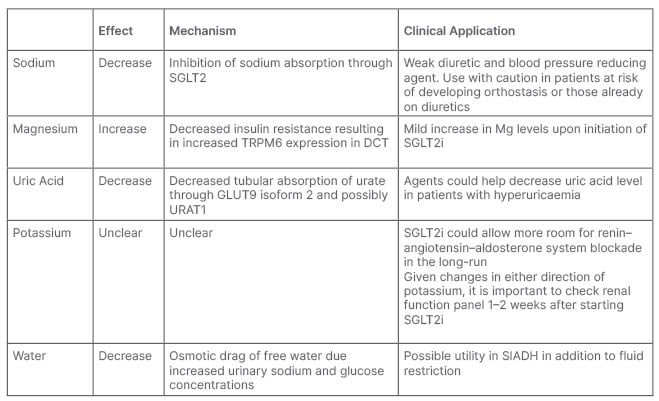
Table 1: Clinical applications of biochemical effects of sodium–glucose co-transporter inhibitors.
DCT: distal convoluted tubules; GLUT9: glucose transporter 9; Mg: magnesium; SGLT: sodium–glucose co-transporter; SGLTi: sodium–glucose co-transporter inhibitor; SIADH: syndrome of inappropriate antidiuretic hormone secretion; URAT1: urate transporter 1.