Abstract
Renal diseases involving glomerular deposits of fibrillary material are an important diagnostic challenge for an ultrastructural pathologist. Several renal diseases are characterised by the presence of fibrillary material in the glomeruli. Two disorders of this type, termed ‘fibrillary glomerulonephritis’ (characterised by fibrils measuring approximately 20 nm in diameter) and ‘immunotactoid glomerulonephritis’ (characterised by larger, microtubular deposits), have been described. The possible relatedness of these two disorders and their potential association with other systemic illnesses are the subjects of current debate. Other multisystemic diseases, including amyloidosis and various forms of cryoglobulinaemia, can also present with fibrillary or microtubular deposits in the kidney.
The distinction between fibrillary glomerulonephritis, immunotactoid glomerulonephritis, and other processes that have similar ultrastructural features are discussed in this review. Recently, both in fibrillary glomerulonephritis and in immunotactoid glomerulonephritis, the presence of a DnaJ homolog subfamily member 9 has been detected. This antigen is not present in amyloidosis and could be involved in the pathogenesis of these diseases. This review will discuss the role and the relevance of this antigen.
INTRODUCTION
Glomerular deposits with fibrillary structure are encountered in several renal disorders. In some diseases, such as amyloidosis, fibril deposition is typically multisystemic and the biochemical nature of the fibrils is well understood. Additional groups of glomerulopathies involving noncongophilic fibrillary deposits have been described and are known as ‘fibrillary glomerulonephritis’ (FGN) and ‘immunotactoid glomerulonephritis’ (ITG).1 Traditionally, the absence of congophilia has been used to differentiate FGN from amyloidosis. Recently, the discovery of the antigen DnaJ homolog subfamily member 9 (DNAJB9) has been detected in patients affected by FGN, regardless of their congophilia,2 and DNAJB9 was the real marker of FGN and ITG more so than congophilia. This fact means that the congophilic properties of organised fibrillary deposits should not be solely relied upon in differentiating FGN from renal amyloidosis. Mass spectrometry and DNAJB9 immunohistochemistry can be useful in making this distinction. Consequently, this study formulated a new distinction among the different types of FGN. Overall, evidence shows that congophobic FGN is significantly more prevalent in females, while the presence of a monoclonal protein on serum protein electrophoresis or immunofixation significantly prevails in the congophilic FGN.
FGN and ITG are uncommon renal diseases characterised by fibrillary amyloid-like glomerular deposits. FGN was first described in 1977 and is characterised by straight fibrils of 10–30 nm thickness with polyclonal Ig deposition.3
RESEARCH METHODOLOGY
Because the aim of this review was to find out what is new in the classification and differentiation between fibrillary and immunotactoid GN, the authors analysed the available papers on FGN and ITG, by a review of the currently available literature. A literature search was performed using PubMed (NCBI/NIH) with the search words “fibrillary glomerulonephritis” and “immunotactoid glomerulonephritis”. As first-line research, the papers published in the last 3 years were examined. Papers were selected according to the relevance of the journal, the authors, the dimension of the study, and the novelty of the findings. In doing so, 20 recently published papers were selected, then the authors proceeded in a reverse chronological order and studies previously published were also included.
EPIDEMIOLOGY AND DISTINCTION BETWEEN FIBRILLARY GLOMERULONEPHRITIS AND IMMUNOTACTOID GLOMERULONEPHRITIS
FGN has been reported to account for 0.5–1.0% of glomerulonephritis, while ITG is 10-fold rarer4 and was first reported in 1992.5 Several authors suggest that fibrillary and microtubular Ig deposits should be considered as variants of the same glomerulopathy, referring the disease to a fibrillary immunotactoid GN.6,7 Other authors highlight that it is essential to distinguish between ITG (microtubular) and FGN.8,9 To date, most authors retain that FGN should be distinguished from ITG on immunopathologic, ultrastructural, and clinical grounds.8,10-12 Table 1 reports the main immunologic and clinical characteristics that distinguish FGN and ITG.
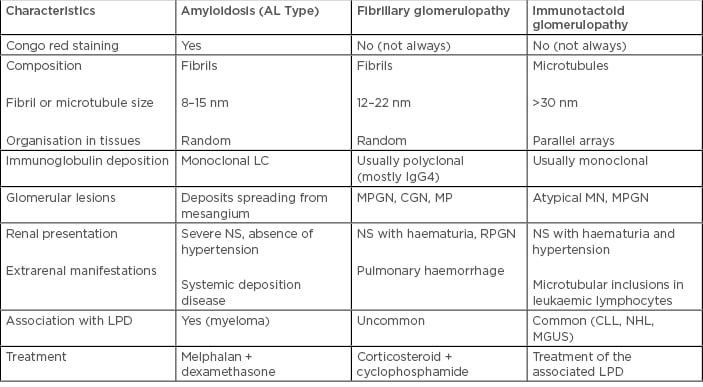
Table 1: Immunologic and clinical characteristics of fibrillary and immunotactoid glomerulopathies.
AL: amyloid light chain; CGN: crescentic glomerulonephritis; CLL: chronic lymphocytic leukaemia; GN: glomerulonephritis; LC: light chain; LPD: lymphoproliferative disorder; MGUS: monoclonal gammopathy of undetermined significance; MN: membranous nephropathy; MP: mesangial proliferation; MPGN: membranoproliferative glomerulonephritis; NHL: non-Hodgkin lymphoma; NS: nephritic syndrome; RPGN: rapidly progressive glomerulonephritis.
Both diseases are most commonly idiopathic, but ITG, in particular, may be associated with monoclonal gammopathies13 and chronic infections.14,15 Haematological malignancies may be frequently found in patients affected by ITG.10 In contrast to FGN, patients with ITG frequently have hypocomplementaemia and an underlying dysprotidaemia. The glomerular deposits are usually monoclonal.16 The differential diagnosis of the two diseases may be easily performed using the algorithm shown in Figure 1.17
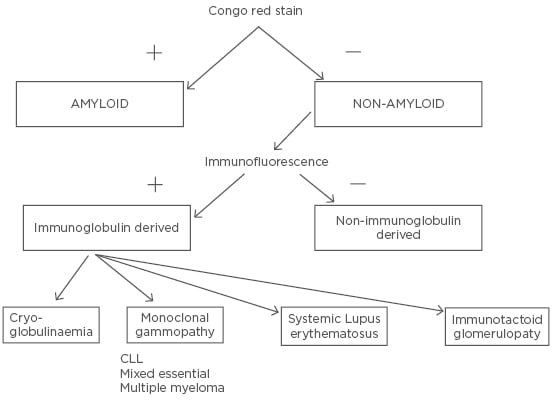
Figure 1: Algorithm for fibrillary glomerulopathy in vitro identification.
CLL: chronic lymphocytic leukaemia.
In a study by Fogo et al.,18 limited by their institution, it was found that patients affected by FGN were significantly younger than patients with ITG. All patients were affected by marked proteinuria and microscopic haematuria, and 70% of patients with ITG had an associated haematopoietic disease with monoclonal proteins and abnormal plasma cell proliferation. ITG is characterised by glomerular deposition of larger microtubular structures (usually >30 nm in diameter) that have focal parallel alignment. In FGN, the microfibrils had a diameter of 14.0±0.5 nm, while the microtubular structures had a mean diameter of 42.3±0.3 nm.
More recently, Lusco et al.19 and Fogo et al.20 identified as characteristic of FGN the following key diagnostic features: 1) mesangial proliferation and variable endocapillary proliferation; 2) polyclonal IgG and complement component 3 (C3); and 3) randomly arranged fibrils in the mesangium and variably in the glomerular basement membrane (GBM), 12–22 nm in diameter and negative for Congo red stain. Similarly, key diagnostic features for ITG were: 1) mesangial proliferation and variable endocapillary proliferation; 2) often clonal Ig and light chain restriction; 3) microtubular in parallel array in the mesangium and variably in GBM, >30 nm in diameter.
The distinction between the two entities is not always easy because many diseases affecting the kidney may present similar structures in the kidney.
This point is well highlighted by a study reporting five cases of patients, in which fibrillary deposition presented in the kidney biopsies.1 The authors stress that the comprehensive analysis of fibrillary glomerulopathies should include a perfect assessment of ultrastructural features (fibril diameter and substructure); exclusion of amyloidosis; correlation between light microscopy, histochemistry, and immunofluorescence; and a careful search for underlying conditions such as lymphoma, chronic disease, and cryoglobulinaemia.
FIBRILLARY GLOMERULONEPHRITIS
FGN was first reported in 19773 as a glomerulopathy with material very similar to amyloid that did not stain with Congo Red, although recently a variant has been identified that is Congo Red positive.20 The term was later changed by Duffy et al.21 who introduced the term ’fibrillary renal deposits and nephritis’. Finally, Alpers et al.22 shortened the name to FGN.
FGN is characterised by the deposition of randomly arranged microfibrils with a diameter from 12 to 30 nm. At immunofluorescence, polyclonal IgG and C3 are present. In light microscopy, several histological patterns may be observed ranging from membranoproliferative GN, to mesangial proliferative GN, to diffuse proliferative GN, or membranous thickening of the capillary tuft.23
For the diagnosis of FGN, the use of electron microscopy is mandatory. Clinically, FGN manifests in proteinuria, often in the nephritic range. The disease course is generally progressive towards end stage renal disease. Fibrils recur in 50% of the transplanted kidneys but the recurrence is often benign.24
The pathogenesis of fibrillogenesis has not yet been elucidated. Fibril deposition is almost always limited to the kidneys, even if some reports describe extrarenal involvement.25 According to other authors, these reports should be interpreted with caution.26 In a series of reported FGN, fibrils co-localise with amyloid P.27 In other series, fibronectin has been detected in the deposits,28 but it appears that fibronectin is not an essential component of the fibrils.
All these findings allowed the improvement of the diagnostic algorithm shown in Figure 1, the updated version of which is shown in Figure 2.
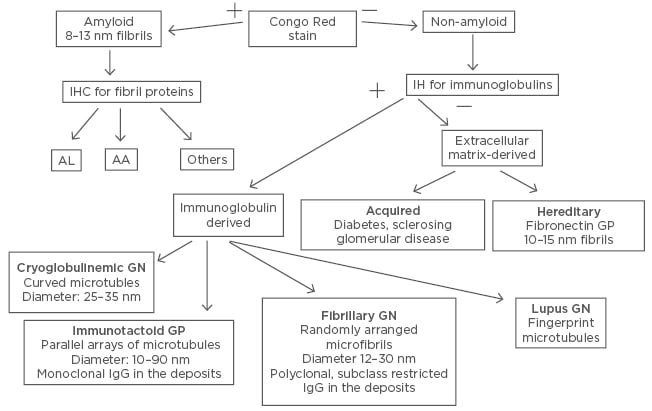
Figure 2: Glomerulopathy with organised deposits identification in vitro.
AA: amyloid associated protein; AL: amyloid light chain; GN: glomerulonephritis; GP: glomerulopathy; IHC: immunohistochemistry.
In a recent study,29 DNAJB9 was detected in patients affected by FGN. The proteome of these patients also contained IgG1 as the dominant Ig. In these patients, immunofluorescence and immunohistochemistry with an anti-DNAJB9 antibody showed a strong and specific staining of the glomeruli, similar to immune deposits. These findings confirmed a previous study30 and identified DNAJB9 as a putative autoantigen in FGN. An immunoprecipitation-based multiple reaction that allows the measurement of serum levels of DNAJB9 has recently been developed. In patients affected by FGN, a 4-fold higher abundance of serum DNAJB9 was found and this suggested that serum levels of DNAJB9 could be a valuable marker to identify FGN.31
A different study20 found that the proteomic signature of amyloid was not detected using mass spectrometry among cases of congophilic FGN. Additionally, DNAJB9 was not detected using mass spectrometry in all cases of FGN, regardless of congophilia, and was absent in cases of amyloidosis and in healthy individuals.
In conclusion, the distinction between FGN and amyloidosis should not be done only on congophilic properties, but by identifying the presence of DNAJB9 with mass spectrometry and immunohistochemistry.
The prognosis of FGN is generally poor, although remission may occur in a minority of patients without immunosuppressive therapy.32
The published experience on the treatment of FGN includes several cases reports using the blockade of angiotensin II and a variety of immunosuppressive treatment including steroids and other immunosuppressants.16
However, no treatment has been shown to improve the long-term outcomes. In a recent study,33 the use of rituximab was reported to treat 10 patients affected by FGN. The study documented the efficacy of rituximab, principally in patients with non-progressive renal failure. A recent study reported the efficacy of Acthar gel in the treatment of FGN.34
IMMUNOTACTOID GLOMERULONEPHRITIS
ITG was first used by Schwartz and Lewis17 to describe a glomerular disease characterised by staining negative with Congo Red and having organised deposits that stain for IgG and complement. It is also defined by the glomerular deposition of microtubules that have distinct hollow centres, which can be 10–90 nm in size.35 Because the deposition in ITG can appear similar to those in cryoglobulinaemia and lupus nephritis, these entities must be ruled out before a diagnosis of ITG can be made. Since the first descriptions, most published series included patients with underlying haematologic malignancies, excluding patients with systemic lupus erythematosus or cryoglobulinaemia.10,11,18
In the study by Rosenstock et al.,4 the incidence of serum or urine monoclonal gammopathy was 67% in patients with ITG, versus 15% in those with FGN. Comparatively, patients with ITG have a significantly higher rate of paraproteinaemia than those with FGN (33% versus 7%, respectively).7
The disease is very rare and the pathogenetic mechanism of fibril deposition is not completely understood. The deposition should possess three features: 1) the Ig found in the deposits must be produced by lymphocytes and/or plasma cells; 2) the Ig precursors must reach the kidney via the circulation; 3) the exclusive glomerular localisation of the deposits implies a role for local factors in fibrillogenesis.
Recent studies on CD2-associated proteins in knockout mice provide the support for a defect in glomerular function in ITG that could be responsible for tactoid formation and suggest that the effect is localised to the podocyte.36,37
Histologically the disease may present as a membranoproliferative GN, a diffuse proliferative GN, or a membranous glomerulopathy. The disease may recur after transplantation. The electron microscope shows microtubular, often parallel arrays with a diameter >30 nm.
The key diagnostic features of ITG are a mesangial proliferation with clonal IgG and light chain restriction on the already described microtubules. Ig is significantly more frequently associated with monoclonal protein and haematopoietic malignancy, most often B cell related. Patients present with nephritic proteinuria, reduced glomerular filtration rate, and hypocomplementaemia. The clinical improvement of proteinuria when treatment was directed at the haematopoietic disorder further support a role for monoclonal proteins in these patients.
Treatment with steroids alone or combined with cyclophosphamide, chlorambucil, or melphalan has been successfully used to induce complete or partial remission of the nephritic syndrome; fludarabine4 or a combination of high-dose methylprednisolone and rituximab followed by alemtuzumab38 has been shown to reduce proteinuria and improve renal function.
With a better understanding of the pathogenesis, clone-directed strategies, such as rituximab against CD20 expressing B cells and bortezomib against plasma cell clones, have recently been used in the treatment of this disease. These clone-directed therapies have been found to be more effective than immunosuppressive regimens used in non-monoclonal protein-related kidney diseases.39
CONCLUSION
The following key diagnostic and clinicopathologic features should be highlighted: the two diseases are distinct and not different aspects of the same disease as previously thought. In both diseases, mesangial proliferation and variable endocapillary proliferation are present. In FGN, randomly arranged fibrils in the mesangium and GBM are present, while in ITG microtubules in parallel arrays in mesangium and GBM are present: such microtubules are larger in diameter. In FGN, polyclonal IgG and C3 are present, while in ITG often clonal IgG and light chain restriction are present. The association with lymphoplasmacytic disorders is uncommon in FGN and common in ITG.