Abstract
Prolonged intermittent renal replacement therapy (PIRRT) is emerging as an alternative to continuous renal replacement therapy as renal support for the critically ill. Unfortunately, dosing data are lacking for many antibiotics, including ceftriaxone. To allow clinicians to prescribe ceftriaxone effectively and safely in this setting, an understanding of the effects of PIRRT on the plasma pharmacokinetics (PK) of ceftriaxone is required. In this case, the authors describe the PK of ceftriaxone in a critically ill patient on PIRRT for the treatment of presumed spontaneous bacterial peritonitis. Blood samples were taken over two dosing intervals: one during PIRRT, and the other off PIRRT. A one-compartment PK model was used to describe ceftriaxone PK; little difference in clearance was noted with and without PIRRT. The authors suggest that ceftriaxone at a dose of 2g qd during PIRRT therapy is adequate to maintain serum levels above the minimum inhibitory concentrations for likely pathogens of non-central nervous system infections in critically ill patients.
INTRODUCTION
Continuous renal replacement therapy (CRRT) is the standard renal support for critically ill patients with acute kidney injury.1 Prolonged intermittent renal replacement therapy (PIRRT) has emerged as an alternative to CRRT because it has lower operating costs while maintaining efficacy, patient safety, and mobility.1 There are various data sources providing advice around antimicrobial dosing for patients receiving intermittent haemodialysis and CRRT, but data are limited for patients undergoing PIRRT and largely confined to case reports.2-⁴ This increases the potential for inadequate or potentially toxic dosing of antimicrobial agents in critically ill patients receiving PIRRT.2
Ceftriaxone is a third-generation cephalosporin with a broad spectrum of antibacterial activity, suitable for the treatment of spontaneous bacterial peritonitis (SBP).⁵ Optimal bactericidal activity is determined by the percentage of time unbound ceftriaxone concentration remains above the minimum inhibitory concentration (MIC) of the pathogen.⁶ For patients with normal renal function, a daily dose of 2 g is able to maintain peritoneal concentrations above the MIC for common SBP causative isolates.⁵ Cephalosporins are predominantly renally excreted and accumulate in patients with renal impairment; however ceftriaxone is an exception because of its high level of protein binding, hepatic metabolism, and biliary excretion.⁵ Despite this, pharmacokinetic (PK) studies of ceftriaxone in patients with normal and impaired renal function have shown a prolonged half-life and decrease in urinary recovery for those with reducing renal function.⁷ The reduction in drug elimination has a non-linear correlation with reducing renal function that may be related to elimination by both renal and non-renal pathways.⁷
Past studies have shown that the unbound concentration and volume of distribution of ceftriaxone is affected by critical illness, resulting in lower trough concentrations, and consideration for decreasing the dosing interval or changing to a continuous infusion were suggested.7,8 However, it has been demonstrated that patients on ceftriaxone requiring CRRT therapy have an equivalent clearance to those with normal renal function and dose adjustments are not required for these patients.⁹,1⁰ Information to guide ceftriaxone dosing for critically ill patients undergoing PIRRT is lacking.
The aim of this report is to describe the PK of ceftriaxone in a patient with SBP undergoing PIRRT and to provide dosing advice for this setting.
METHODS AND RESULTS
Patient Characteristics
A 44-year-old male (height: 164 cm; weight: 148 kg) with a history of Child–Pugh B hepatic cirrhosis, ascites, congestive cardiac failure, atrial fibrillation, diabetes, and chronic kidney disease was diagnosed with SBP. He received an ascitic paracentesis for fluid overload, resulting in drainage of 14 L of fluid, which was complicated by acute-on-chronic renal impairment and was therefore transferred to an intensive care unit for dialysis and inotropic support. The ascitic fluid was negative in the bacterial culture and ceftriaxone was commenced empirically for SBP treatment. His serum albumin was 33 g/L on the day PIRRT commenced.
Antibiotic Dose and Administration
Because the recommendation from the literature is that patients on ceftriaxone requiring CRRT or intermittent haemodialysis therapy do not require dosage adjustments,⁵-11 the patient received a dose of 2 g intravenously over 30 minutes qd for both PIRRT and non-PIRRT cycles.
Prolonged Intermittent Renal Replacement Therapy
PIRRT (Fresenius 5008, Fresenius, Sydney, Australia) was conducted as a 10-hour treatment in the haemodiafiltration mode with a heparinised circuit and a 1.4 m2 filter (Ultraflux® AV600S, Fresenius), with the following renal replacement therapy settings: blood flow rate 200 mL/min; dialysate flow rate 200 mL/min; and ultrafiltration flow rate 250 mL/h, except for a 1-hour period where the ultrafiltration flow rate was 275 mL/h. The transmembrane pressures ranged from 50 to 135 mmHg. Fluid removal per ultrafiltration was 1,964 mL in 10 hours on the day of sampling. PIRRT was initiated on Day 1 of the study period for just under 10 hrs.
Blood Sampling
PIRRT was commenced after the patient received their first dose of ceftriaxone and blood was sampled at 4.5, 8.5, 11.5, and 16.5 hours after dosing for this PIRRT session. Blood samples were also taken at 2.0, 5.5, 9.0, and 12.0 hours after dosing for the non-PIRRT session which occurred after the patient’s second dose of ceftriaxone. A total of eight samples were taken throughout a 2-day study period which included one PIRRT session. Of the four samples taken for the PIRRT session, PIRRT was active for the 4.5 hour and 8.5 hour samples, and was ceased prior to taking the remaining two samples. Samples were stored at -80°C prior to assay at Pathology Queensland Laboratories, Brisbane, Australia. The dialysate was not assayed to assess PIRRT clearance of ceftriaxone.
Drug Assay
Serum unbound ceftriaxone concentrations were measured by validated ultra performance liquid chromatography coupled with QDa mass detection (Waters Corporation, Milford, Massachusetts, USA) at Pathology Queensland. Unbound concentrations were separated using a validated ultrafiltration method with an Amicon® Ultra 0.5 mL 30,000-molecular-weight-cutoff centrifugal filter device (Merck Millipore, Sydney, Australia). The assay had a lower limit of quantification of 0.1 mg/L and the imprecision was <10% across three quality-control levels.
Pharmacokinetic Modelling
PK data was analysed using a non-parametric method with library package for R, Pmetrics™ (Laboratory of Applied Pharmacokinetics and Bioinformatics, Hollywood, California, USA) with testing of both one- and two-compartment models. A second clearance term representing the presence of PIRRT was tested and excluded due to mathematical insignificance. Both additive (λ) and multiplicative (γ) error models were tested. Inspection of the log-likelihood ratio and goodness-of-fit models was used to select the final model.
Consent
Informed consent was obtained to collect blood samples and report this case.
Pharmacokinetics and Pharmacodynamics
As both one- and two-compartment models produced similar parameters, the one-compartment model was selected on the principle of parsimony. The clearance of ceftriaxone was 5.23 L/hr for this patient and the estimated volume of distribution in the central compartment was 27.07 L. Because PIRRT did not have a clear effect on clearance there is no separate PIRRT clearance value. The intercompartmental rate constant from peripheral to central compartment (KPc) and central to peripheral compartment (KcP) were 6.95 h-1 and 8.24 h-1 respectively.
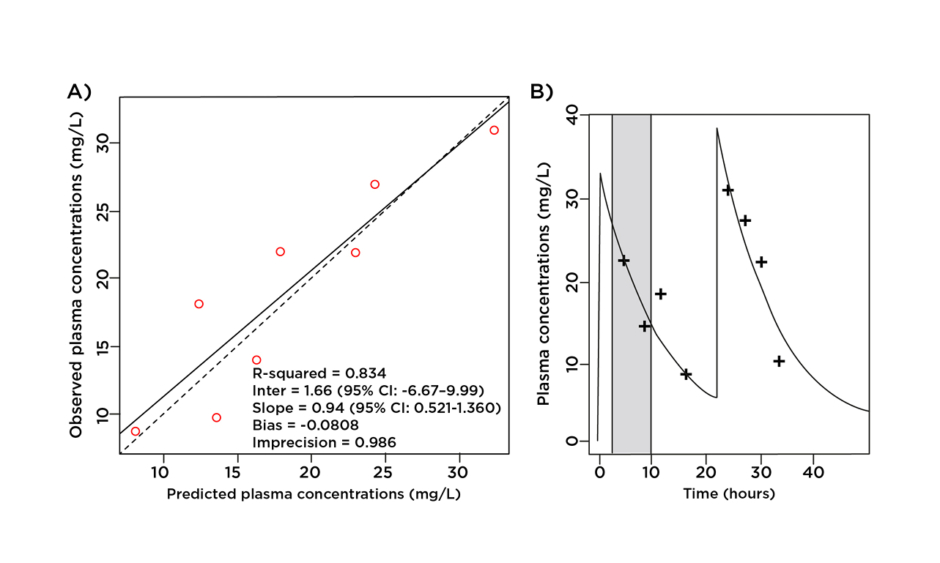
According to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) and Clinical & Laboratory Standards Institute (CLSI), the MIC breakpoint for ceftriaxone for the treatment of Enterobacteriaceae is 1 mg/L.12,13 As displayed in Figure 1, an intravenous dose of 2 g qd ensured unbound ceftriaxone plasma concentrations well above this breakpoint for the entire dosage interval while undergoing PIRRT.
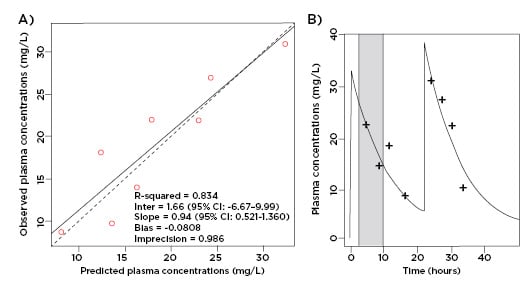
Figure 1: A) Observed-predicted plot for ceftriaxone plasma concentrations (mg/L); B) predicted unbound plasma concentration time profile versus observed data. Prolonged intermittent renal replacement therapy is shown in grey.
Clinical Outcome
The patient was successfully discharged from the intensive care unit but subsequently died from respiratory failure due to ascites and fluid overload.
DISCUSSION
To the best of the authors’ knowledge, this is the first report of the PK of ceftriaxone in a patient receiving PIRRT. Data are limited on describing the PK of antimicrobials, which are highly protein bound and predominantly nonrenally eliminated in patients receiving PIRRT.
Anidulafungin is a highly protein-bound (84%) echinocandin antifungal which is degraded in human plasma with minimal renal excretion of unchanged drug.1⁴ When given to an adult undergoing PIRRT, the PK data were comparable to those of healthy adults without renal dysfunction, suggesting that dose adjustment is unnecessary.1⁵
The fluoroquinolone moxifloxacin has moderate protein binding of approximately 54%, is predominantly hepatically metabolised, and also has minimal urinary excretion.1⁶,1⁷ In a study of moxifloxacin PK during PIRRT, the mean volume of distribution and half-life were similar in critically ill patients requiring PIRRT and in healthy subjects.1⁶ Standard dosing of 400 mg qd given post-PIRRT was recommended.1⁶
The lincomycin clindamycin is also highly protein bound and hepatically cleared, and does not require dosage adjustment during CRRT.⁷ Drug–protein complexes have a large molecular weight and are not removed by CRRT as readily as drugs with a low protein binding capacity.⁷
The above data suggest that for moderately to highly protein-bound drugs with predominantly nonrenal clearance, such as ceftriaxone, PIRRT also does not readily remove drug–protein complexes. This may explain the lack of change in ceftriaxone clearance in our patient in whom albumin levels were within the normal therapeutic range while on PIRRT despite having a history of Child–Pugh B cirrhosis and ascites, which could further alter ceftriaxone PK.
An increase in free ceftriaxone concentration after PIRRT was observed and may demonstrate a redistribution of ceftriaxone into the vascular system. This effect has been previously described during plasma exchange and would be expected to affect highly protein bound drugs.1⁸ Accumulation of ceftriaxone may result from multiple doses of ceftriaxone during PIRRT cycles, however this is an area that requires further research.
CONCLUSION
In this patient, the clearance of ceftriaxone was not substantially affected during PIRRT, and ceftriaxone dosage adjustments should not be required to achieve PK or pharmacodynamic targets for the treatment of SBP. The authors believe this to be the first reported case on the PK of ceftriaxone in a patient with Child–Pugh B hepatic cirrhosis, ascites, and chronic kidney disease requiring PIRRT, so further studies involving larger patient numbers are required to confirm this recommendation.