Abstract
Several diseases are related to complement involvement. In particular, its role is essential for the pathogenesis of several renal disease. On the other hand, the complement role may also be protective, and this possibility should be well known when managing complement inhibitors.
Complement inhibitors are relatively newly discovered therapies that are essential in some diseases, and in others improve the efficacy of the already known therapeutic measures that represent the standard of care.
In the case of glomerular diseases, complement plays a role in almost all diseases. In some diseases, complement abnormalities represent the prevailing factor in the pathogenesis. In such diseases, complement inhibition represents an essential therapy. In other diseases, complement plays an important role, but other factors are involved in the pathogenesis. Clearly, in these diseases, complement inhibition represents a therapy that could be added to the standard of care therapy, according to the physician’s judgement. Examples of these diseases are lupus nephritis, thrombotic microangiopathy when associated to some cases of lupus nephritis, and IgA nephropathy. The latter is one of the first primary glomerulonephritides in which a relevant role of complement is documented. These three diseases are the object of this brief review. Particular concern should be given to ongoing clinical trials. Indeed, many of these anti-complement therapies are still in different phases of clinical trials. Finally, particular concern must be ascribed to the problem of associating these emerging therapies to already existing and proven treatments.
Key Points
1. The complement system is involved in several renal diseases. In some disease it is involved deeply, and in such diseases the treatment is principally based on anti-complement drugs. In other glomerular diseases, the complement system is involved marginally.
2. In some renal diseases, principally in autoimmune diseases, the complement system may have a protective role. Indeed, the early complement components have an effect on opsonisation and clearance of immune complexes, apoptotic cells, and cellular debris. Additionally, genetic deficiency of complements 1 and 4 leads to a higher risk of having lupus nephritis. A different protective role exerted by complement is the enhanced immune complexes clearance generated by complement 3b when bound to completment receptor 1 on the erythrocyte surface.
3. Problems in targeting complement in lupus nephritis include which pathway to target, as classical, lectin, and alternative pathways may be involved; which immunosuppressant to use, with a choice between steroids, mycophenolic acid, calcineurin inhibitors, and rituximab; and compatibility of complement therapy with standard of care. Steroids and complement inhibitors are synergistic, while calcineurin inhibitors are synergistic with complement inhibitors. Complement inhibitors may reduce the effect of rituximab.
INTRODUCTION
The complement system has been an appealing drug target for a long time.1,2 However, early drug development efforts have failed for two reasons: the lack of specificity of the anti-complement drugs, and the insufficient knowledge about the mechanism of action of complement in both health and disease.
COMPLEMENT SYSTEM ABNORMALITIES
Complement system abnormalities may be due to alterations of the different complement proteins, or of the protein system related to complement regulations. Often, these abnormalities are on a genetic basis, and abnormalities frequently involve factor H. They may consist of factor (f) H mutation or presence of anti-fH autoantibodies. Less frequent is a mutation of factor I or of the membrane cofactor protein. Abnormalities of the complement system may concern gain of function of complement (C) 3 or of complement fB. These abnormalities are also on genetic basis.
To date, it is well known that complement is deeply involved in several renal diseases. Such diseases should be divided according to the relevance of complement in their pathogenesis. In several diseases, complement exerts a principal and fundamental role, such as in atypical haemolytic uremic syndrome (aHUS) and in C3 glomerulonephritis. In such diseases, different treatments may be used, ranging from non-specific immunosuppression to newer complement targeting agents. Anyway, such patients should receive anti-proteinuria therapy with angiotensin converting enzyme inhibitors. Non-specific immunosuppression includes steroids, rituximab (RTX), and mycophenolate mofetil (MMF).3,4 In other diseases, such as systemic lupus erythematosus (SLE); secondary thrombotic microangiopathy (TMA), often associated with lupus nephritis (LN); and several glomerulopathies, such as IgA glomerulonephritis, the role of complement according to new knowledge is particularly relevant, and the anti-complement therapies are more often associated to the standard of care therapies. In this short review, the involvement of complement in diseases such as secondary TMA, SLE, and IgA nephropathy (IgAN) will be treated, looking at the main targeting drugs.
In other renal diseases, complement is similarly involved, even if at a lesser degree, including antineutrophilic cytoplasmic antibody vasculitis, membranous nephropathy, and diabetic nephropathy.5 First, it should be highlighted that complement activation is not only restricted to the glomeruli within the kidney, but several experimental and clinical data6-9 have indicated that complement activation may contribute to tubular cell injury. However, it should be observed that some of these studies have been conducted on animals.
An important consideration is that complement has not only a damaging effect, but may also have a protective role against autoimmune diseases. Table 1 documents that the deficiency of some complement proteins leads to disease. In the human, the genetic deficiency of C1 has a 77% risk for SLE, and the genetic deficiency of C4 has a 75% risk for SLE.10 Indeed, the early complement components have an important effect on the opsonisation and clearance of immune complexes, apoptotic cells, and cellular debris, which are all important in the initiation and pathogenesis of LN.
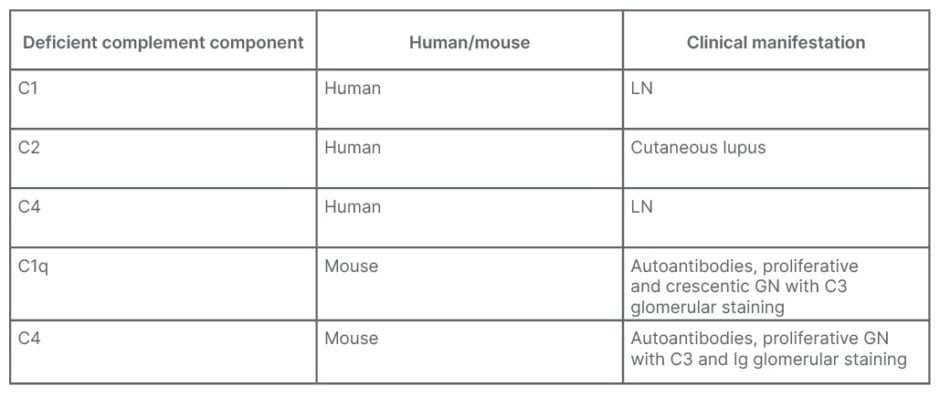
Table 1: Complement may have a protective role against autoimmune diseases.
C: complement; C1q: complement component 1q; GN: glomerulonephritis; LN: lupus nephritis.
A different protective role exerted by complement is the enhanced immune complexes clearance generated by C3b when bound to complement receptor 1 (CR-1) on the erythrocyte surface. Indeed, immune complexes activate the complement system, and the generated C3b binds to the complexes and to CR-1 present on the surface of erythrocytes. During erythrocyte traffic through sinusoids in liver and spleen, residual phagocytes remove bound immune complexes, leading to their clearance.11-13
Several recent studies using transcriptomics techniques to identify complement proteins in kidney biopsies have provided documentation of an increase of messenger RNA of complement proteins in patients with SLE. During inflammation, there is an increase of such proteins, as well as a decrease of messanger RNA of complement regulators such as CR-1.
Through this way, Tempe et al.14 identified an accelerated intrarenal synthesis of distinct classical and alternative complement pathway components, most associated with impaired renal function. The study documented that glomerular complement synthesis is associated with interferon signalling, while tubulointerstitial complement synthesis is associated with aberrant T cell receptor signalling.
Studies from Parikh et al.15,16 using transcriptome analysis found that complement molecular profiling of kidney compartments from renal biopsies differentiated responders to treatment and patients affected by lupus flare. These findings may help as a treatment guide and to predict responses better.
Similarly, complement system activation studied by transcriptome analysis allowed identification of complement protein activation in chronic antibody-mediated rejection, and in IgAN recurrence.17 In summary, all these studies on complement regulation and kidney diseases allow the statement that complement activation in nephrology has a double-edged role.18
Standard kidney biopsies may be useful in understanding intrarenal complement activity, but the use of kidney biopsies with complement staining or transcript analysis adds useful information in understanding the exact role of complement in a particular patient. The use of such techniques may allow moving toward a personalised approach in the management of LN.19 The study by Gilmore et al.20 using transcriptome analysis on formalin-fixed paraffin-embedded kidney biopsy tissue revealed which molecular pathways are active in each patient, with LN comporting clinical utility in treatment selection. Similarly, Liu et al.,21 studying kidneys of patients with LN and New Zealand Black/White mice, found higher expression of C3 and TGF-β1 in the earlier phases of LN. Mejia-Vilet et al.22 found a significant heterogeneity in immune gene expression in the kidney of patients with LN. This fact might have relevance in treatment decisions. In a similar study, Peterson et al.23 characterised heterogeneity in the molecular pathogenesis of LN from transcriptional profiles of laser-captured glomeruli.
Finally, Martin et al.24 found that plasma C4d correlates with C4d deposition in kidneys of patients affected by active LN, and Wang et al.6 found that membrane attack complex (MAC)deposition in renal tubules is associated with interstitial fibrosis and tubular atrophy.
Tubular complement activation and deposition occurs in proteinuric diseases, and in LN, C5-C9 deposition is associated with tubulointerstitial fibrosis. In addition, there is a subgroup of patients with persistent subclinical inflammation. Such patients have elevated biomarkers (monocyte chemoattractant protein-1) associated with a lower kidney survival.
TARGETING COMPLEMENT IN LUPUS NEPHRITIS
To date, several questions are open in the issue of targeting complement in LN, including which pathway to target; which combination of immunosuppressants to use; when to add anti-complement therapy to standard of care treatment; in which phase of renal disease complement should be targeted; and which new anti-complement compounds to use for LN.
Which Pathway to Target?
Figure 1 shows that all three pathways of complement activation are involved in LN: the classical pathway, the lectin pathway, and the alternative pathway.
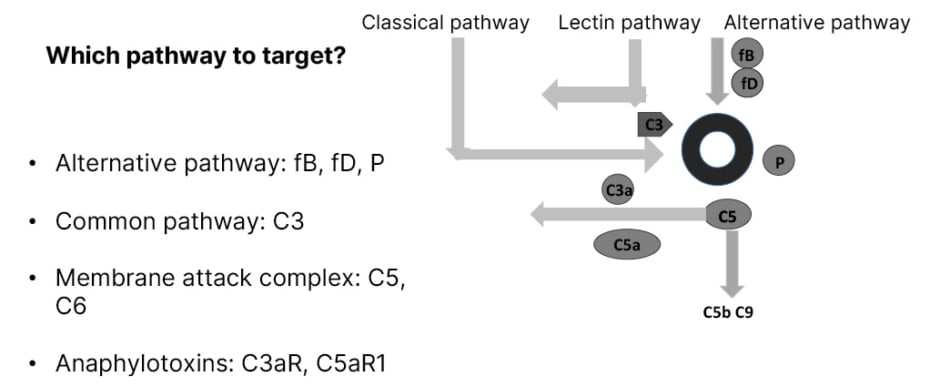
Figure 1: Complement as a therapeutic target therapy in lupus nephritis.
C: complement; fB: factor B; fD: factor D; P: properdin; R: receptor.
A main rule is to avoid the inhibition of the early classical pathway in order to maintain the immune complexes clearance. In the alternative pathway, targets are fB, fD, and properdin. In all pathways, C3 is a possible target. Other targets are the MAC and the anaphylotoxins C3a receptor and C5a receptor.
Which Immunosuppressant?
The second question is which is the best immunosuppressant to target complement. A study from Lemercier et al.25 documented that glucocorticoids modulate the monocyte secretion of C3, fB, and fH, indicating an anti-inflammatory property and a control of complement activation.
A study from Wang et al.,26 done on animals, documented that a compound of mycophenolic acid, in addition to targeting the proliferation of lympholeukocytes, downregulates complement protein 3 and other pro-inflammatory cytokines, such as TNFα-, IL-6, and TGF-β.
An old study on animals, from Pennington et al.,27 documented that cyclophosphamide was able to inhibit the synthesis of C2 and C4 in a guinea pig model. A study from Renner et al.,28 conducted on mice, documented that the treatment of patients with calcineurin inhibitors (CNI) is associated with an increased production of complement-activating endothelial micro particles. The deposition of C3 on the surface of endothelial micro particles was examined by flow cytometry. Complement activation caused by cyclosporine-induced micro particles seems to be due to a decreased binding of factor H.
In a different study,29 CNI-induced complement activation downregulated the suppressor of cytokine signalling and the complement inhibitors CD46 and CD55. According to these studies, complement activation may contribute to chronic calcineurin inhibitor nephrotoxicity. However, it is only a hypothesis that cyclosporine causes alternative pathway dependent injury in the kidney and vasculature. Moreover, many of these studies were conducted on animals. Additionally, large recent studies have documented the efficacy of CNI in the treatment of LN.30
The effect of RTX on LN has been discussed and its efficacy is still controversial.31 Studies on patients with B cell malignancies treated with RTX32 did not document RTX efficacy in targeting complement.
Compatibility of Complement Therapy with Lupus Nephritis Standard of Care
According to some studies, several complement proteins are downregulated by corticosteroids,33 while other studies document an increase of C3 and fB messanger RNA.25 Overall, the administration of a complement inhibitor to glucocorticoids could synergise with complement inhibition. MMF reduces C3 expression,26 and the addition of a complement inhibitor could enhance the effect of MMF.
The cited study on cyclophosphamide27 documents its inhibition on C2 and C4, which has a negative effect on the immune complexes clearance, but the addition of a complement inhibitor acting on a late stage of complement pathway may synergise with the anti-inflammatory effect of cyclophosphamide. CNIs enhance the inflammatory effect of complement in the kidney.
The use of a complement inhibitor could reduce this effect, allowing the use of CNIs in the treatment of LN.
RTX depletes B cells through different mechanisms, among which the complement-dependent cytotoxicity is relevant.34 The addition of a complement inhibitor to RTX could reduce its effect on B cells through complement-dependent cytotoxicity.
In Which Phase of Renal Disease Should Complement Be Targeted?
Four phases should be distinguished in the development of LN. The first phase is autoimmunity, in which there is loss of self-tolerance; the second phase consists of the asymptomatic immune complexes deposition; the third phase is the inflammatory response that corresponds to clinical LN; and the fourth phase is the repair with development of fibrosis that corresponds to chronic kidney disease. The use of complement inhibitors is more effective when given to patients with active LN with clinical inflammation. The main problem is understanding when this phase occurs. It is best to have biomarkers corresponding to intra-renal complement activation, independent from biomarkers corresponding to systemic complement activation.
Transcriptomic analysis of kidney biopsies documented elevated expression of genes associated with C3 and fD during renal inflammation.35 Other studies using transcriptomic analysis documented high expression of fB, fH, and properdin in patients with active LN documenting the activation of the alternative pathway.36 In addition, urine examination may be useful in detecting intrarenal inflammation. Patients with active LN also have urinary C3d37 and elevated urinary levels of MAC.38 In conclusion,5 the renal diseases in which alterations of complement system are present more frequently are aHUS, C3 glomerulonephritis (C3GN), IgAN, and LN.
In aHUS, the most frequent abnormality is the one concerning fH, but less frequently, abnormalities of MCP or fI have been found, and abnormalities of fB or C3 have been encountered with consequent gain of function.
In C3GN, all the described abnormalities can be found, but the more frequent abnormality is the presence of C3 nephritic factor autoantibodies that stabilise C3 convertase.
In IgAN, the abnormal IgA activate the complement system through the alternative or the mannose-binding lectin pathway. In LN, data on humans have documented abnormalities of fB.
Overall, patients who could benefit from anti-complement therapy, looking at the Mayo Clinic Study,4 are patients with lower serum creatinine at the beginning, less proteinuria at diagnosis, less extensive glomerulosclerosis at renal biopsy, and with a lesser degree of tubular atrophy and interstitial fibrosis.
Complement Targeting Drugs
Dysalarm-322 is a drug still on clinical trial39 that aims to investigate the role of anti-C1s, anti-high mobility group box 1 protein, and anti-C1q autoantibodies in the pathogenesis of LN. Previous studies40 have already documented the role of such autoantibodies. Eculizumab, a monoclonal antibody (mAb), principally used for the treatment of aHUS, has also been used for the treatment of LN resistant to other therapies.41 Ravulizumab, like eculizumab, is a novel humanised mAb that targets C5, and is under investigation for proliferative LN and IgAN. Narsoplimab is a human mAb acting on the lectin pathway as it binds to the serine protease 2 (MASP-2) that activates the lectin pathway. A Phase II clinical trial is underway to evaluate its efficacy on LN.42 Iptacopan targets fB and is active on the alternative pathway. To date, a Phase II trial evaluates the efficacy and safety of iptacopan (LNP023) in combination with standard of care for active LN Class III and IV.43 Pegcetacoplan (APL-2), an inhibitor of C3, is to date being evaluated in several nephropathies, including LN.
Finally, other compounds could have an effect on LN. These compounds are to date the object of clinical trials for other glomerulopathies, but are not yet studied for the treatment of LN.44
TARGETING COMPLEMENT IN PATIENTS WITH LUPUS NEPHRITIS AND ASSOCIATED THROMBOTIC MICROANGIOPATHY
Patients with LN and associated TMA should be managed according to the underlying aetiology of TMA, as stated by the Kidney Disease Improving Global Outcomes (KDIGO) guidelines 2021.45 Patients with this syndrome should be distinguished according to having low ADAMTS13 levels; normal ADAMTS13 levels and negative antiphospholipid antibodies; or normal ADAMTS13 levels, but positive for antiphospholipid antibodies. The first group of patients should be treated by plasma exchange, glucocorticosteroids, RTX, and caplacizumab (an anti-von Willebrand factor).46 Patients with normal ADAMTS13 levels should be screened for complement-mediated TMA and treated by eculizumab if positive. Finally, patients positive for antiphospholipid antibodies should be treated like patients with antiphospholipid syndrome nephropathy.
TARGETING COMPLEMENT IN PATIENTS WITH IgA NEPHROPATHY
Among the different glomerulonephritis, pathogenesis of IgAN is deeply involved by complement activation and/or dysfunction.47
IgAN is characterised by deposition of immune complexes containing polymeric galactose-deficient IgA1 in the glomerular mesangium. Polymeric IgA1 and IgA1-containing immune complexes can activate the alternative and lectin pathway, leading to cleavage of intact C3, thereby forming C3a and C3b. C3a is an anaphylotoxin and C3b activates the complement cascade. In addition, fH is a key regulator of the complement system and together with fI, fH cleaves C3b to the inactive form iC3b. In IgAN, fH-related proteins could compete with the regulatory functions of fH, thereby promoting complement activation.
Due to the strong association between glomerular complement activation and IgAN, complement inhibitors are being tested in several clinical trials. Table 2 shows the principal clinical trials to date involved in testing complement inhibitors for the treatment of IgAN.48-55
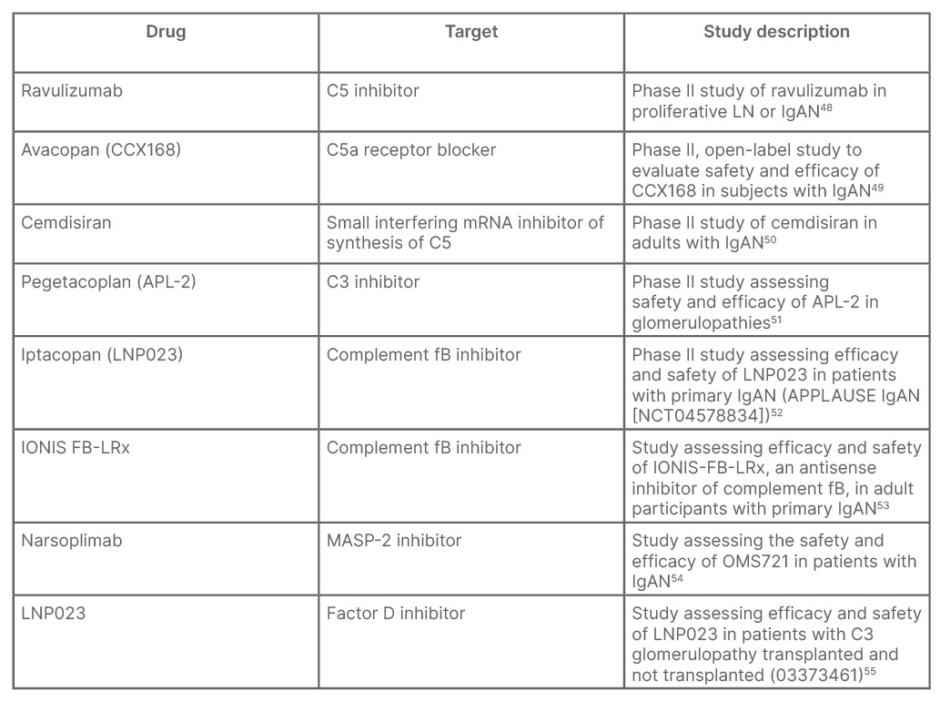
Table 2: Principal studies to test efficacy of complement inhibitors in IgA nephropathy.
fB: factor B; IgAN: IgA nephropathy; LN: lupus nephritis; MASP-2: mannan-binding lectin serine protease 2; mRNA: messenger RNA.
C5a receptor 1 is a complement receptor that regulates the dendritic cells, and is essential to T cell immunity. Ravulizumab, a humanised mAb anti-C5 prevents cleavage of C5 into C5a and C5b. To date, a Phase II study of ravulizumab in proliferative LN or IgAN (SANCTUARY)48 has the objectives to evaluate the safety and efficacy of ravulizumab administered by intravenous infusion compared to placebo, and demonstrate proof-of-concept of the efficacy of terminal complement inhibition in participants with LN (LN Cohort) or IgAN (IgAN Cohort). Preliminary data have documented its efficacy in reducing proteinuria and improving the estimated glomerular filtration rate (eGFR).
Another oral C5a inhibitor, avacopan (CCX168; NCT02384317),49 is the object of a pilot study to test its safety, tolerability, and efficacy in reducing proteinuria in patients with IgAN and persistent proteinuria, despite supportive therapy with a maximally tolerated renin-angiotensin-aldosterone system blocker.
Cemdisiran (ALN CC5) is a synthetic, small RNA inhibitor designed to suppress liver production of C5, which may reduce terminal complement pathway activation and subsequent inflammation. A Phase II, double blind controlled study (NCT03841448)50 aims to evaluate the safety and efficacy of cemdisiran in patients with IgAN.
APL-2 is a peptide inhibitor of C3. A Phase II trial will assess the safety and preliminary efficacy of daily APL-2 subcutaneous infusion administered for 16 weeks with a 6-month safety follow up, in patients with glomerulopathies, including IgAN.51
Serum levels of fB are increased in patients with IgAN, correlating with the activation of B cells. Blocking fB has potential pathophysiological importance. IgAN proteinuria reduction after 90 days of treatment with LPN023 (a fB inhibitor) is being tested in a Phase IIa/IIb trial (NCT03373461).52 IONIS-fB-LRx is an antisense inhibitor of complement fB. It is to date the object of a Phase II study to evaluate the effectiveness and safety in adults with primary IgAN (NCT04014335).53
Narsoplimab is a human mAb against MASP-2. MASP-2 is essential for the activation of the lectin pathway that contributes to disease progression of IgAN and is, therefore, a potential drug target. Narsoplimab has been designed to treat diseases mediated by the lectin pathway of complement through inhibition of MASP-2. A first Phase II study was designed to assess the safety and efficacy of narsoplimab (OMS721) in patients with IgAN (NCT03608033).54 BCX9930 is a fD inhibitor. Its efficacy in C3G, IgAN, and primary membranous nephropathy is evaluated in a long-term study (NCT05162066).55
Lafayette et al.56 published a study evaluating the effects of narsoplimab on patients affected by IgAN. The study concluded that narsoplimab is safe and well tolerated, with a clinical meaningful reduction in proteinuria, and stability in eGFR in high risk patients with advanced IgAN. Similarly, LPN023 is evaluated to control IgAN.57
Several of the studies have been the object of pre-emptive publication, including one on ravulizumab,58 two on iptacopan,59,60 one on avacopan,61 and four on narsoplimab.62-65 In a pilot study by Bruchfeld et al.,66 the C5a receptor inhibitor avacopan in IgAN documented the improvement of eGFR.
WHICH TREATMENT AFTER KIDNEY TRANSPLANTATION?
It is difficult to give an answer that is valid for any nephropathy. It depends on the type of kidney disease, and the severity of the disease itself.
In the case of LN, the risk of recurrence ranges between 2–30%.67 In most cases, mild histologic lesions characterise recurrence and only rarely does recurrence lead to graft failure. The post-transplant immunosuppression is probably sufficient to control the disease. Generally, it can be stated that the result of kidney transplantation in LN depends on the clinical conditions at transplantation.
Suarez et al.68 conducted a study on the recurrence and graft loss of patients affected by C3GN. The lowest incidence of graft loss was in patients treated with eculizumab. Among those who received no treatment because of a stable graft function, the allograft loss was 32% in patients with C3GN, and 53% in patients with dense deposit disease.
No treatment is needed for patients with IgAN, while the problem for patients with aHUS is still not solved, even if many patients try to interrupt the treatment, slowly and with a careful control.
CONCLUSION
In conclusion, the author has several points as final remarks, but several problems still remain to be solved. The current developments show that complement inhibition in renal disease is actively pursued in several clinical studies. The technological advances and clinical experience with eculizumab have led to a new confidence in therapeutic strategies that target the complement system.
The expanding list of trials and the increasing number of complement inhibitors, which are being developed and are tested in preclinical studies, demonstrate that complement inhibition is an option for therapy of glomerular disorders. Given the pathological heterogeneity between, and even within, indications for complement specific therapies, careful patient stratification will be essential to pave the way toward new therapeutic options.
Still unsolved problems are as followed: as aforementioned, for each single renal disease it is possible to target complement in different ways with different drugs and different targets. One problem is deciding which target should be chosen for any disease. A different problem is choosing which inhibitors to choose when having different inhibitors for the same target. Finally, a further problem is to understand how long the anticomplement therapy should be maintained. This point has a particular relevance in consideration of the high cost of such therapies.