Meeting Summary
This 1-day meeting was held at the Austria Trend Savoyen Hotel, Vienna, Austria. The morning session consisted of a series of presentations by experts in the field of cystinosis. Prof Katharina Hohenfellner gave an overview of nephropathic cystinosis (NC) and provided her own experience of a multidisciplinary patient follow-up. Dr Alexey Tsygin provided his long-term experience of treatment with cysteamine and highlighted the importance of treating early and the role of cysteamine in treating and preventing long-term complications of cystinosis. Dr Roser Torra presented the findings of the T-CiS.bcn project, which aimed to develop a transition plan from paediatric to adult care through a multidisciplinary team. Dr Rachel Bishop provided an overview of the ocular manifestations of cystinosis followed by a presentation on the most recent data with gel-like Cystadrops® from Dr Hong Liang.
The key themes from the morning sessions were:
- With the development of specific treatment for cystinosis, the disease paradigm has shifted from a paediatric illness where patients suffer end-stage renal disease (ESRD) by 10 years of age to a longerterm multisystemic illness with extra-renal complications
- Earlier treatment of NC with cysteamine can delay both the renal and extra-renal manifestations of the disease
- There are difficulties in transitioning from paediatric to adult care and this can lead to reduced treatment adherence; a multidisciplinary team may help to smooth the transition
Cystinosis: Prevalence, Burden, Complications, Diagnosis, Prognosis, and Management
Professor Katharina Hohenfellner
Cystinosis is a very rare autosomal lysosomal storage disease with an incidence of approximately 1:100,000–200,000 live births. Cystinosis is caused by biallelic mutations in the CTNS gene which maps to chromosome 17p13. More than 100 mutations have since been described and most lead to complete loss of cystine transporter function which is required to transport free cystine from the lysosome to the cytoplasm. Consequently, when cystinosin is deficient, cystine accumulates in lysosomes where crystals are formed, leading to progressive organ damage.1
Three clinical types of cystinosis can be distinguished dependent on age at presentation and symptoms: infantile NC, late-onset NC, and ocular cystinosis. Infantile NC is the most frequent (95%) and the most severe form of cystinosis. Asymptomatic aminoaciduria, a first sign of proximal tubular damage, is present after birth. Clinical symptoms generally appear by the age of 12–24 months in the form of polyuria, polydipsia, dehydration acidosis, hypokalaemia, rickets, and failure to thrive as result of renal Fanconi syndrome (which consecutively leads to progressive loss of glomerular function and ESRD, requiring renal dialysis and transplant).
Patients with infantile NC often survive until an average age of 10 years without cystine-depleting therapy and renal-replacement therapy. However, with the introduction of cysteamine bitartrate,2 the only treatment available for patients with infantile NC, survival increased and allowed patients to reach adulthood, potentially reaching >60 years of age.
Cysteamine bitartrate was introduced in 1994 in the USA and 1997 in Europe. Cysteamine delays progression to ESRD by 6–10 years and has also been shown to postpone extra-renal complications. Patients require life-long therapy, especially after kidney transplantation, and can experience side effects, such as gastrointestinal symptoms (e.g. nausea and vomiting) and halitosis. In patients treated with high-dose cysteamine, skin striae, bone pain, myalgia, and endothelial proliferative lesions on the elbows (reactive angioendotheliomatosis) have been reported. However, these side effects are reversible after reducing the dose of cysteamine.
Postnatal screening provides early diagnosis and enables the start of treatment which in turn would delay the onset of ESRD. Postnatal screening however, is not widely available.
Systemic treatment does not influence corneal cystine accumulation; life-long local application is necessary to reduce photophobia and prevent severe corneal involvement.
As patients are surviving longer, multisystemic disease with extra-renal complications (due to cystine accumulation in other organs) is becoming apparent. The most commonly affected organs and systems are the eyes (corneal crystals and retinal degeneration leading to blindness), neuromuscular (facial muscle weakness, swallowing difficulty, distal vacuolar myopathy, and gait disturbance), endocrine and bone (hypothyroidism, diabetes mellitus, delayed sexual maturation in males, and poor growth), and pulmonary systems (extra-parenchymal restriction of ventilation secondary to myopathy).3-5
In 2012, a model for comprehensive interdisciplinary treatment was established through collaboration of the Traunstein Hospital, Traunstein, Germany, and a German patient support group. The primary objectives were to build a multidisciplinary team that would address all the organs involved and to create a transition model for patients with a very rare multisystemic disease. In order to accomplish these objectives, both adults and children are seen by a range of 14 specialists, including an ophthalmologist, dermatologist, nephrologist, and endocrinologist within a 6-hour period. During this time, clinical and laboratory analyses are conducted. This comprehensive care is provided once a year, in addition to patients’ regular medical care. The team treat and manage both children and adults and are therefore able to better monitor the course of disease and can therefore introduce preventive strategies as a focal point of treatment.
The interdisciplinary Cystinosis Clinic Traunstein, Traunstein, Germany, has seen 75 patients since it was initiated in 2012. The data of a subgroup of 20 adult patients in primary nephrological care indicated that they were all experiencing severe extra-renal complications. Concerning cysteamine treatment, 3 out of 20 patients had been without medication for a period longer than 5 years. Ophthalmological involvement was present in all 20 patients and all presented with photophobia. Cystadrops were used by 12 patients on a regular basis. Within the subgroup, 5 out of 20 patients presented with retinopathy, correlating with insufficient systemic treatment. The muscle status was found to be unsatisfactory in 13 of the 20 patients; 5 patients developed restrictive lung disease and 3 patients had severe swallowing problems. Gastrointestinal symptoms were present in half of the patients. These late complications have been found to be related to cysteamine dosage (i.e. patients who stopped cysteamine treatment were more likely to have extra-renal complications). However, presence of halitosis has not shown any indication of being related to dosage.
Considering that cysteamine therapy was introduced 30 years ago, it is mandatory that a careful evaluation of the first adult generation be conducted in order to obtain a clear and detailed picture of the long-term benefits and drawbacks of this form of treatment.
Outlining the Benefits of Long-Term Treatment with Cysteamine: Sharing Data
Doctor Alexey Tsygin
A case was presented of a female aged 34 months, who was the first case of NC in Russia. She had insufficient growth and weight gain. At the age of 1 year she presented with polydipsia, polyuria, and vomiting. She was found to have rickets, glycosuria, phosphaturia, and low serum levels of potassium, phosphate, and bicarbonate. She underwent routine treatment with vitamin D but showed no improvement. At the age of 1.5 years she was found to have creatinine elevation and anaemia. On ultrasound, she had enlarged kidneys with no cysts and no nephrocalcinosis. Corneal deposition of cystine crystals was also observed and treatment with cysteamine was commenced.
The patient’s symptoms were typical of early findings in cystinosis, which included Fanconi syndrome, rickets, corneal crystals, glomerular crystals, abnormalities of the tubular epithelial cells on electron microscopy, and ‘swan neck’ deformities.5 Eleven years later the patient is still short, with some bone deformities, but can attend school, has clear corneas, and has a glomerular filtration rate of 58 mL/min, indicating that renal function should be preserved for several years without the need for renal replacement therapy.
The recommended cysteamine dose is 1.30 g/m2/ day in children up to 12 years, and 2 g/day in children older than 12 years and over 50 kg in weight, divided into four doses.2 White blood cells are used for measurement of cystine levels. Normal levels are <0.2 nmol hemicystine/mg protein. In untreated patients with cystinosis, levels may be 2–15 nmol hemicystine/mg protein, while with adequate treatment cystine levels should be ≤1 nmol hemicystine/mg protein; levels should be checked 5–6 hours after cysteamine intake. Adherence to treatment is difficult to maintain in children; the use of gastrostomy to provide parenteral nutrition and medication may be successful for increasing height and body mass. In addition, the adverse events (AEs) associated with cysteamine treatment, such as diarrhoea, vomiting, and the smell are a barrier to adherence, particularly in adolescents.
Early initiation of treatment and good adherence are important for achieving good long-term outcomes. If cysteamine treatment is started before the age of 2.5 years it significantly decreases the velocity of the rise in serum creatinine and improves growth rates.6 When cysteamine treatment is taken regularly with a sufficient dosage and white blood cell cystine levels are kept low, growth almost approaches normal levels; however, growth is poorer in patients receiving only partial treatment or no treatment.7 Similarly, creatinine clearance is better in patients receiving regular cysteamine treatment.8 Likewise, ESRD has also been shown to be delayed when mean leukocyte cystine levels are kept low,9 when treatment is started early (<5 years of age),10 and when overall adherence to treatment is good.9 In addition, the healthcare system plays a role; renal survival is poorer in developing countries with no cysteamine treatment available compared with countries where patients receive cysteamine treatment.11
Data from the European Society for Paediatric Nephrology (ESPN) and European Renal Association and European Dialysis and Transplant Association (ERA-EDTA) registry from 1979–2008 have shown that the age at start of renal replacement therapy has increased with the advent of cysteamine treatment, rapidly increasing from 1990, while similar increases have not been observed in patients starting renal replacement therapy for non-NC indications.12 In addition, prognosis appears to be better in patients who have a renal transplant for NC than in patients who have a transplant for other reasons;12 this may be because patients with NC who have received a transplant now have a functioning cystine transporter and are able to clear cystine from their kidneys normally,13 so primary disease does not recur to the kidney but remains systemic. With the improved outcomes for patients with NC, patients may now suffer from longer-term extra-renal complications of cystinosis. Cysteamine treatment has been shown to prevent or reduce these complications, including Type 1 diabetes mellitus, myopathy, pulmonary dysfunction, hypothyroidism, and death.1
Question and Answer Session
During the Question and Answer (Q&A) session, the potential for NC screening was raised. Prof Hohenfellner advised that there are currently no screening tools available; the leukocyte numbers are too low in the dried blood spot to permit screening. Dried blood spot screening would be ideal as there is already the healthcare infrastructure in place to support it. Within her centre, they are evaluating the possibility of urine dipstick screening. Dr Tsygin recommended that all siblings of an affected patient should be genetically screened, although acknowledged that this may raise some ethical issues, especially for prenatal diagnostics.
Cystinosis: Exploration of Best Practice Recommendations/Guidance in Both Paediatric and Adult Care
Doctor Roser Torra
The fact that patients with NC are surviving longer means there has been a paradigm shift from considering NC as a paediatric disease primarily affecting the kidney, to an adult chronic, multisystemic disease. This is associated with new challenges in disease management including adherence to life-long treatment and the transition from paediatric to adult care. In paediatric care, patients receive personalised and permanent attention from physicians and family, which is challenging to integrate into regular adult care follow-up. The transition from paediatric to adult care is poorly co-ordinated, and approximately 35% of young adults can lose a successful kidney transplant within 36 months of transition.13
The wellbeing of adolescents with cystinosis is affected by multiple disease-related factors including long-term disabilities, multiple hospitalisations, gastrointestinal complications, photophobia, myopathy, and delayed puberty, which can in turn influence their choice of profession, relationships, pursuit of family life, and their friendships. Therefore, it is important to maintain the best possible control of the disease to reduce burden and improve quality of life.
The T-CiS.bcn project was developed to improve the continuum of care with a guided transition from paediatric to adult cystinosis services while increasing patient empowerment in moving from guardian-care to self-care. The objective was to develop a consensus document with recommendations for each speciality for the transition of paediatric to adult care and to propose a transition scheme.14
The project found that there should be a thorough management of the transition process. The transition plan should be designed and started when the patient is pre-adolescent (12–15 years), with training during adolescence (16–18 years), and transfer during young adulthood (19–25 years) (Figure 1). The main specialists who look after the patient may change with age; for instance patients in infancy and childhood will need genetics and pathophysiology, gastroenterology, and nephrology, while in adolescence and adulthood they may need transplant specialists and have psychosocial concerns etc.
Figure 1: Timeline of transition from paediatric to adult care.
The solution to the varying needs of the patient throughout the transition proposed by the T-CiS.bcn project is the multidisciplinary team, which is composed of different specialists with complementary skills and knowledge working together to make treatment recommendations that facilitate quality of care. The team aims to address the whole clinical management of the physical and psychological needs of the patient. Visits are co-ordinated among the specialists, and a joint action protocol facilitates management and transition. The team has regular meetings to discuss cases, protocols, and projects, and the case manager nurse is essential to its smooth co-ordination.
Questionnaires as part of the project identified several barriers, fears, and feelings about the transition. Within paediatric care patients have strong relationships with the team, it is a safe environment with an organised level of care led by the paediatric nephrologist; however, within adult care the rare diseases may be unknown to adult specialists and the care is not as personalised, with gaps in resources, and patients may be transferred without an agreed plan. For patients and families, there is a wish for autonomy, although still to be connected to a care team, and a fear of discontinued care. Paediatric specialists expressed concern regarding the absence of reference centres and expert professionals and noted a requirement for better communication with the adult specialist and comprehensive care after the transition, and a need for a co-ordinated and planned transition.
Recommendations from the T-CiS.bcn project team for improving treatment adherence include: identification and management of risk factors (e.g. patient and socio-economic factors, disease-related factors; treatment-related factors and healthcare system organisation barriers); identification and assignment of a patient co-ordinator; promotion of patient education and treatment support with disease education programmes; therapeutic plans that are easy to follow with support measures for therapeutic compliance; use of questionnaires to detect non-compliance; and follow-up of appointments and absences; development of a patient support programme involving family members, friends, and patient associations; creation of a multidisciplinary team; and implementation of protocols to manage the transition from paediatric to adult care.15
Other recommendations for guiding the transition include advising the patient when they reach the transition age and activating the protocol in an individualised way, applying referral centre selection criteria, identifying the most suitable adult nephrologist, establishing communication between the case manager at the paediatric hospital and the counterpart at the adult receiving service, elaboration of a concrete transition plan and ensuring that the pharmacy service adapts and that the patient has sufficient medication until they are incorporated into the new centre.
Question and Answer Session
During the Q&A session the speakers discussed the practicalities of running a multidisciplinary team. It may not be possible to have paediatricians present in all meetings together with adult specialists if they are not part of the same centre. A multidisciplinary team setting may be able to improve adherence to treatment.
Ocular Complications of Cystinosis
Doctor Rachel J. Bishop
Deposition of cystine crystals in the eye leads to development of ocular symptoms.16 One of the main ocular manifestations of cystinosis is photophobia, which can often be severe. It usually appears by 3–4 years of age, but is often not reported until later. All structures of the eye can contain crystals; crystals in the cornea and retina/retinal pigment epithelium are the most significant in terms of the patient complaints to the ophthalmologist. Secondary effects of the crystals include increased intracranial pressure, which can lead to optic nerve damage. Corneal crystals are initially evident by approximately 1 year of age; they may start in the periphery and move towards the centre and posterior of the eye. In later, untreated cases, there may be crystals in the entire peripheral and anterior central stroma. When they are severe, crystals are present throughout the stroma, and the cornea has a hazy, ground glass appearance. However, they may have a minimal effect on visual acuity.
Other symptoms may include foreign body sensation, band keratopathy, severe dry eyes, filaments, and limbal neovascularisation. Long-term complications may include thickening and transillumination of the iris, and development of posterior synechiae, which cause scarring impeding normal fluid movement, and may lead to angle closure glaucoma and phthisis, and formation of a pupillary membrane with crystals. Posterior ocular complications relating to crystals in the retina include a characteristic peripheral retinopathy that may be recognisable from birth. It is initially peripheral but progresses centrally as the child ages if untreated. The optic discs and retinal vessels are spared but the optic nerve may be involved due to papilloedema from raised intracranial pressure. Retinal degeneration is associated with impaired visual function, as measured by colour vision, visual fields, and electrodiagnostic tests.
The retina is vascularised and therefore systemic cysteamine treatment is able to reach the eye and improve retinal symptoms. Corneal symptoms are managed by eye drops as the cornea does not have a blood supply. Other treatments in severe cases may include ethylene diamine tetra-acetic acid (EDTA) chelation and corneal transplant for band keratopathy. However, following corneal transplant, crystals may reaccumulate due to the infiltration of host inflammatory cells into the donor cornea. In patients with angle-closure glaucoma surgery to open the iris and potentially cataract surgery may be used, and in the case of papilloedema, shunts may be used to drain fluid from the eye, thus releasing the pressure.
Ocular non-NC is uncommon; patients do not develop renal failure, and have crystals in the cornea and bone marrow. The only symptom is photophobia and patients exhibit retinal pigment abnormality. They carry one severe and one mild mutation on the CTNS gene.
Ocular cystinosis has been recognised for a long time,17,18 with studies from up to three decades ago demonstrating the potential for removal of corneal crystals by topical cysteamine treatment.19-21 The main reasons for the effectiveness of topical versus oral treatment include the fact that topical treatment permits higher concentrations of cysteamine to reach the eye, and in addition, the cornea has no blood supply, and therefore oral cysteamine cannot reach the stromal cells.16 Cystaran™ was first approved in the USA in 2012 and in Europe in 2017 and are effective (Figure 2).21 However, frequent dosing is needed (every hour while awake); therefore it is difficult to maintain adherence to treatment. Altered treatment regimens may be useful; a study found that by altering the timing of the doses e.g. by grouping some doses together, adherence could be improved; although this may be limited by the tolerability.22
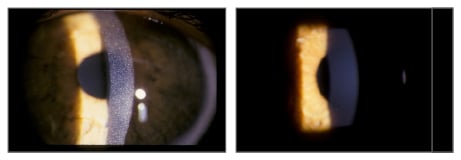
Figure 2: Corneal cystine crystals before and after topical cysteamine treatment.
Taking time in the early appointments to teach the patient how to use the eye drops, and starting with a manageable goal in terms of frequency of dosing and increasing gradually are useful strategies for achieving good adherence. Patients should visit the ophthalmologist at least yearly, and visits should be tailored to how the patient is responding to the treatment. It is important for patients to stay connected with other patients and families and to be aware of ongoing research. Future goals may be to develop a less arduous treatment regimen, to investigate means of preventing crystal formation, and to develop a cure for the illness.
Ophthalmology Latest Data with Gel-Like Cysteamine Eye Drops
Doctor Hong Liang
Two clinical trials have investigated the new gel-like cysteamine eye drop formulation. OCT-1 was an open label, single-group, Phase I/II safety study of 8 patients treated over 5 years.23 CHOC was an open label, randomised, comparative parallel group Phase III trial of 32 patients treated over 3 months in two centres in France, which aimed to determine the superiority of the gel-like drop 0.55% (n=15) compared with the usual hospital pharmacy preparation 0.10% (n=16).24
Within the CHOC trial there were a number of clinical assessments including clinician-assessed photophobia, visual acuity (LogMAR scale), visual contrast sensitivity scale, corneal staining total score, corneal cystine crystal score, intraocular pressure, and especially the new developed high resolution imaging technology such as in vivo confocal microscopy (IVCM), and anterior segment optical coherence tomography (OCT). After 3 months of treatment, visual acuity and intraocular pressure were the same between groups, while improvements were observed in the visual contrast sensitivity scale and the corneal staining total score. Significant improvements were observed in clinician-assessed photophobia, corneal cystine crystal score, and anterior segment OCT. Significant improvements were also demonstrated in IVCM total score (Figure 3) and confirmed in each corneal layer.
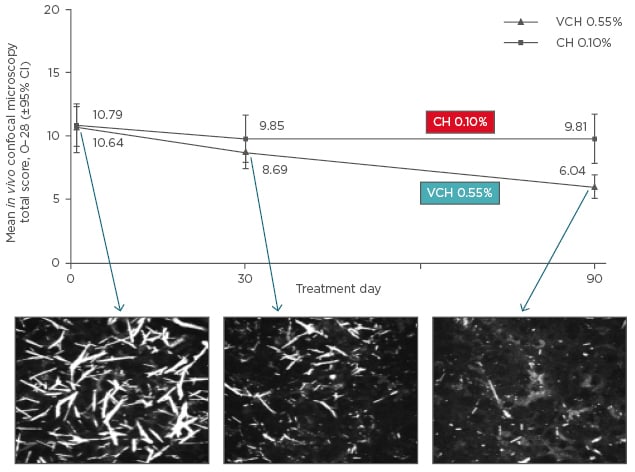
Figure 3: Change in in vivo confocal microscopy total score from baseline to Day 90.
CI: confidence interval; VCH: Cystadrops®; CH: control cysteamine hydrochloride eye drop formulation.
Improvements in IVCM total score were accompanied by reductions in photophobia and corneal cystine crystal score.
There were no severe AEs during the trial; AEs that occurred more frequently in the patients receiving gel-like drops included stinging, redness, burning, blurred vision, and itching. Overall, the gel-like formulation given 1–4 times a day was efficacious and safe in paediatric and adult patients with cystinosis and corneal cystine crystal deposits.
There are still some key points to understand with regard to ocular cystinosis. Firstly, when neovascularisation and band keratopathy are present it may not be possible to save the cornea; this may be a problem of the limbus, and corneal crystals may be seen in the limbus using IVCM. Could there be a deficiency of stem cells in the limbus? The therapy strategies implicating stem cell activation may be possible in the future. Secondly, ocular surface disease is still an issue. Deposits of cystine crystals may be seen in the conjunctiva, and issues such as dry eye and meibomian gland dysfunction are becoming hot topics in ocular cystinosis. Inflammation of the conjunctiva may be an issue. In the Q&A session it was highlighted that inflammation can affect adherence to treatment, and therefore there is a need to decrease inflammation, potentially with the use of non-steroidal anti-inflammatory drops or cyclosporine. Thirdly, there are few choices for imaging the retina. Although optic nerve oedema may be observed by using Angio-OCT for the optic nerve observations, the mechanisms underlying the oedema are unknown.