Meeting Summary
Prof Alexander Rosenkranz and Prof Markus Ketteler welcomed the audience and the expert panel of the symposium, and briefly described the programme of the meeting. Prof Laurent Juillard discussed the challenges faced in achieving phosphate control in patients on haemodialysis, as well as aspects for optimising the management of hyperphosphataemia. Prof Philip Kalra described recent advances in hyperphosphataemia treatment, concentrating on an iron-based, calcium-free phosphate binder that may offer a lower pill burden compared with previous treatments, and thereby address the challenge of patient non-adherence.
Hyperphosphataemia: Understanding the Challenges
Professor Laurent Juillard
In healthy individuals, the kidney is the primary regulator of phosphorus homeostasis. When renal function is preserved, there is a balance between the absorption and urinary excretion of phosphate, together with an equilibrium between bone formation and resorption. A net of 800 mg of phosphate from the diet is being absorbed in an exchangeable phosphate pool consisting of three components: 70% comprises the intracellular phosphate involved in phosphorylation reactions (e.g. signalling); 29% comprises the mineralisation of the skeletal system (bones and teeth); and <1% includes serum phosphate that is usually measured for monitoring purposes. The skeletal component is often ignored due to the daily bone formation/resorption, however the skeleton functions as a phosphate reservoir containing >80% of total body phosphate. This reservoir assimilates phosphate when the bone formation balance is positive.1 In chronic kidney disease (CKD), impaired renal function (i.e. reduced urinary excretion) results in reduced phosphate excretion and phosphorus homeostasis is lost. Despite reduced gastrointestinal (GI) absorption, excess of bone resorption compared to formation and accumulation of phosphorus in soft tissue and vasculature lead to an overall positive balance of phosphorus in the body and serum, resulting in hyperphosphataemia.1
As shown by a large epidemiological trial (N=25,529), serum phosphorus is associated with a significant increase in all-cause (5,857 events) and cardiovascular (CV; 1,930 events) mortality risk in dialysis patients.2 However, haemodialysis patients prescribed phosphate binders show a significant reduction in mortality (25% lower death rate, without taking into account nutritional factors). Without adjusting for nutritional indicators, mortality rate was higher in patients not administering phosphate binders (versus those prescribed a phosphate binder) at all serum concentrations ≥3.5 mg/dL. Similar patterns were observed when nutritional indicators were adjusted for.3
Elevated serum phosphate levels are generally associated with an increased risk of CV disease among patients with and without kidney failure. Hyperphosphataemia occurs in dialysis patients when the glomerular filtration rate is reduced, the dialysis is inadequate, and when the patient is not adhering to dietary restrictions or dialysis regimes. This results in direct effects such as vascular injuries due to, for example, induction of oxidative stress and endothelial dysfunction, and indirect effects. This includes modifications in blood parameters such as increased fibroblast growth factor-23 (FGF-23) leading to increased left ventricular hypertrophy;4 inhibition of 1,25-dihydroxyvitamin D synthesis leading to decreased cardiac contractility; arterial calcification; myocardial fibrosis; and increases in parathyroid hormone. The result of this is: cardiac fibrosis, inflammation, and impaired myocardial energy production (Figure 1). All these effects increase CV risk in haemodialysis patients.5
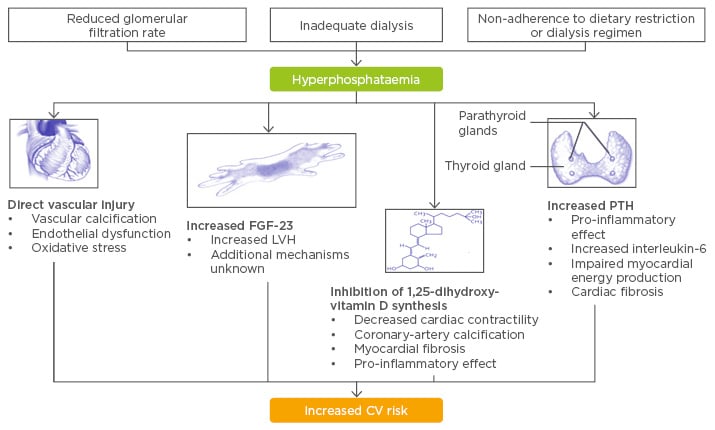
Figure 1: Putative mechanisms linking hyperphosphatemia and cardiovascular disease.
CV: cardiovascular; FGF-23: fibroblast growth factor-23; LVH: left ventricular hypertrophy; PTH: parathyroid hormone.
Adapted from Tonelli M et al.5 and Faul C et al.4
Abnormal phosphate metabolism is also a major mechanism of vascular calcification. The factors that increase vascular calcification include: failed anti-calcific processes (loss of inhibitors); matrix degradation via elastolysis with metalloproteinases; apoptosis (phosphate inflammation); osteochondrogenic differentiation of vascular cells; and remodelling of bone, leading to circulating nucleation complexes, and paracrine factors contributing to initiation or progression of vascular calcification.6
Renal osteodystrophy is a disorder of bone remodelling and is associated with hyperphosphataemia. Insufficient levels of parathyroid hormone characterise low turnover osteodystrophy, where movement of calcium and phosphate from the exchangeable pool into the skeleton is decreased and reductions in bone formation exceed the reductions in bone resorption. As a result, the excess bone resorption contributes to hyperphosphataemia and is illustrated through patients with low turnover osteodystrophy who develop osteoporosis in CKD. In high turnover osteodystrophy, because of the impact of excess parathyroid hormone, high levels of receptor activator of nuclear factor kappa B ligand (involved in osteoclast differentiation) develop, resulting in excess osteoclast activity. Even though bone formation rates are increased, bone resorption rates are excessive, which also contributes to vascular calcification and stiffness.1 Hyperphosphataemia further affects bone health, which in turn affects mortality. Fractures are 17-times more frequent and occur up to 15 years earlier in patients with Stage 5 CKD than in an age-matched general population.7 In addition, there is a >2-fold increase in mortality risk associated with hyperphosphataemia compared with patients on dialysis who do not have hip fractures.8
As mentioned, increased FGF-23 levels as a result of hyperphosphataemia may lead to heart failure. Additional modifications in levels of vitamin D and parathyroid hormone are also contributors to such consequences, particularly in patients on dialysis. The effects however, can be reversed after kidney transplantation.9 Significantly increased levels of FGF-23 in patients with CKD increase the left ventricular mass, leading to a considerable remodelling of the heart, reduction in ejection fraction,4 and inducing mortality in end-stage renal disease.10
The management of hyperphosphataemia faces several challenges, the first of which is nutrition. Caution should be exercised when attempting to restrict the amount of phosphorus in the diet of patients so as to not lead to malnutrition; however, more attention should be paid to the quality of food as many processed foods have a much higher phosphorus content compared with their fresh form. Phosphorus additives are particularly common in frozen foods (72%), dry food mixes (70%), packaged meat (65%), bread and baked goods (57%), soup (54%), and yoghurts (51%).11
The second challenge in the management of hyperphosphataemia includes the effect of dialysis modalities on phosphate removal from the body: conventional haemodialysis, peritoneal, nocturnal, and daily dialyses all remove significant quantities of phosphorus from the blood.12 Phosphorus magnetic resonance spectroscopy during haemodialysis in anephric pigs was used to measure the concentration of intracellular phosphate and concentration of adenosine triphosphate, since inorganic phosphate has an impact on cellular energy.13 The results showed a linear decrease of urea during the dialysis, corrected for acidosis, and calcium in the effluent was almost identical to the levels contained in the dialysis solution. No significant modification in calcium balance and the fact that bone resorption involves more calcium than phosphorus transfer, suggested that the bone was not involved as a source of phosphorus in dialysis. During dialysis, sustained removal of phosphate was observed over 180 minutes. Extracellular content decreased quickly, then slowly plateaued, and increased before the end of dialysis. Intracellular phosphate was shown to increase at the same time as extracellular phosphate plateaued (the inorganic phosphate to phosphocreatine ratio increased by a very significant 6.9% [p<0.00001]).13 At the same time, adenosine triphosphate content inside the cell was shown to decrease significantly during the dialysis and was accompanied by a constant increase in intracellular pH.13 These findings pose the question of whether there is a detrimental cellular effect due to the removal of phosphate during dialysis. This will be assessed in patients with CKD and the outcome is anticipated to influence how dialysis will be performed in the future.
An additional challenge in managing hyperphosphataemia lies with phosphate binders. Their efficacy at lowering serum phosphate levels has been shown to be significant in controlled trials,14-16 however, data from observational studies as opposed to that taken from a clinical setting show 52% of patients achieved phosphorus concentrations within the Kidney Disease Outcomes Quality Initiative (KDOQI) target range and only 27% of patients had a phosphorus concentration within the Kidney Disease: Improving Global Outcomes (KDIGO) target range.17
Haemodialysis patients endure one of the highest pill burdens reported for any chronic disease, with phosphate binders comprising half of a total which can come to 19 pills per day.18 A systematic review of online databases that analysed the prevalence and determinants of non-adherence to phosphate binding medication in patients with end-stage renal disease showed that adherence varied from 22–74% in a given population; the wide range is attributed to different methods of non-adherence assessment (serum phosphate levels [58% adherence] versus self-report measures [31%]).19
In conclusion, Prof Juillard reaffirmed that phosphorus homeostasis is lost in CKD, where impaired renal function leads to a loss of phosphorus excretion resulting in inevitable hyperphosphataemia in end-stage disease. He repeated that hyperphosphataemia is a risk factor for CV disease, vascular and other soft-tissue calcification, bone disease, and increased mortality, and that the management of hyperphosphataemia through dialysis, diet, and phosphate binders is still far from optimal and better treatment options are needed.
Advances in the Management of Hyperphosphataemia
Professor Philip Kalra
As previously described, the mortality rate is very high in dialysis patients. At 25 years of age, men or women on dialysis have around 100-times the risk of CV death versus those in the general population. The gap narrows over time but evens out by the age of 60 or 70 years, when the risk is 10 to 20-fold that of the general population for the same age.20 There are several aetiological factors for structural CV disease, some of which include: hypertension, renin-angiotensin-aldosterone upregulation, inflammation, intra-dialytic ischaemia, oxidative stress, and anaemia. However, CKD and mineral bone disease play a dominant role incausation of vascular disease and uraemic cardiomyopathy. The prevalence of vascular calcification is very high: vascular calcification is found in around 40% patients with CKD new to dialysis21 and in up to 83% of patients on established dialysis.14 The greater the degree of calcification in a patient, the higher the mortality risk: the patients with the greatest calcification have only ~20% all-cause survival at 6 years, compared with 95% survival rate of those with no calcification.22
A study in a USA population by Block et al.23 showed that there is a U-shaped observational association between serum phosphate concentration and mortality in patients with hyperphosphataemia. As serum phosphate concentration increases above 4.0 mg/dL to 5.0 mg/dL, the risk of mortality increases in a stepwise pattern, until at >9.0 mg/dL, the risk of death is twice as high as at 3.1–4.0 mg/dL. The U-shape is attributed to a group within the dialysis population that are often malnourished and have an increased risk of mortality. At the same time, also in the USA population, haemodialysis patients (25%) within the highest range of FGF-23 levels had a nearly 6-times greater risk of mortality compared with those in the lowest quartile.10 Faul et al.4 have shown that injecting FGF-23 directly into the murine myocardium causes gross left ventricular hypertrophy in just 1 week; this important animal work ties in with the impact that hyperphosphataemia has on the human body.
Randomised controlled trials investigating phosphate binder intervention in CKD mineral bone disease range from those looking at vascular calcification (Renagel in New Dialysis [RIND] and Treat-to-Goal), or mortality (Dialysis Clinical Outcomes Revisited [DCOR] and RIND follow-up) as endpoints. The RIND study (N=110) included patients who were incident to dialysis and randomised to sevelamer or calcium-based phosphate binders (CCPB) over 18 months.24 The Treat-to-Goal study involved twice as many patients as RIND, but patients were again randomised to sevelamer or CCPB, and looked at the median percentage change in calcification in the coronary artery and in the aorta.14 Both studies showed a marked attenuation of calcification in patients treated with calcium-free phosphate binders compared to CCPB.
Including over 2,000 patients on dialysis, the DCOR study is the largest phosphate binder study focussing on mortality and compared patients treated with CCPB to those on sevelamer. The primary outcome measure was all-cause mortality and this was not found to be significantly different between the treatment groups. However, in an apparently pre-specified sub-analysis involving patients >65 years of age, sevelamer therapy resulted in a statistically significant reduction of 22% (p=0.03) in the relative risk for all-cause mortality.25 The RIND trial follow-up arm extended to 66 months, during which a significant reduction in mortality was observed amongst patients treated with sevelamer. Mortality rates were 10.6/100 patient years (95% confidence interval [CI]: 6.3–14.9) in subjects treated with calcium containing phosphate binders and 5.3/100 patient years (95% CI: 2.2–8.5, p=0.05) in subjects treated with sevelamer.26
In addition, a meta-analysis of 18 studies investigated the effects of CCPB versus non-calcium phosphate binders (NCCPB) on all-cause mortality. Eleven of those studies were randomised and reported an outcome of mortality (4,622 patients; 936 deaths). A 22% reduction in all-cause mortality was observed amongst patients on NCCPB compared with those on CCPB. Over all the 18 studies, NCCPB were associated with a 13% reduction in all-cause mortality versus CCPB (risk ratio: 0.87, 95% CI: 0.77–0.97), which was irrespective of patient dialysis status.27
As described previously, the pill burden of phosphate binders in CKD patients is very high, leading to a reduction in treatment adherence. The DOPPS study showed that that the more frequently patients skipped their pills, the more likely they were to have serum phosphate levels higher than the target range of 5.5 mg/dL.28
Sucroferric oxyhydroxide (SFOH, [Velphoro®]), is a stabilised polynuclear iron oxyhydroxide-based phosphate binder consisting of iron, sucrose, starch, and water. The iron(III)-oxyhydroxide strongly binds the phosphate by replacing hydroxide groups, giving it its phosphate-binding property, while adding sucrose to iron-oxyhydroxide prevents it from ageing and maintains its phosphate-binding capacity. In a preclinical adenine rat model, SFOH, lanthanum, and sevelamer NCCPB were studied. All three attenuated carotid and aortic calcification (including abdominal, inferior, and superior thoracic aorta calcification) to similar degrees compared with control.29 A Phase III trial investigating the efficacy and safety of SFOH involved over 1,000 patients. After a washout period of 2–4 weeks in patients who were on phosphate binders, patients were enrolled if their serum phosphate levels were in excess of 5.5 mg/dL. Patients were then randomised to either SFOH or sevelamer, in a ratio of 2:1. The starting dose for SFOH was 1.0 g per day, which is two tablets, while for sevelamer it was 4.8 g per day, which is six tablets. The patients were then followed for 24 weeks, followed by a longer phase extending to 52 weeks.30,31
During the Phase III study, non-inferiority of SFOH versus sevelamer carbonate was established; SFOH and sevelamer carbonate showed a comparable phosphate-lowering effect from baseline to Week 24, with a mean daily tablet number of 3.1 and 8.1, respectively: a significant difference in pill burden (Figure 2).30
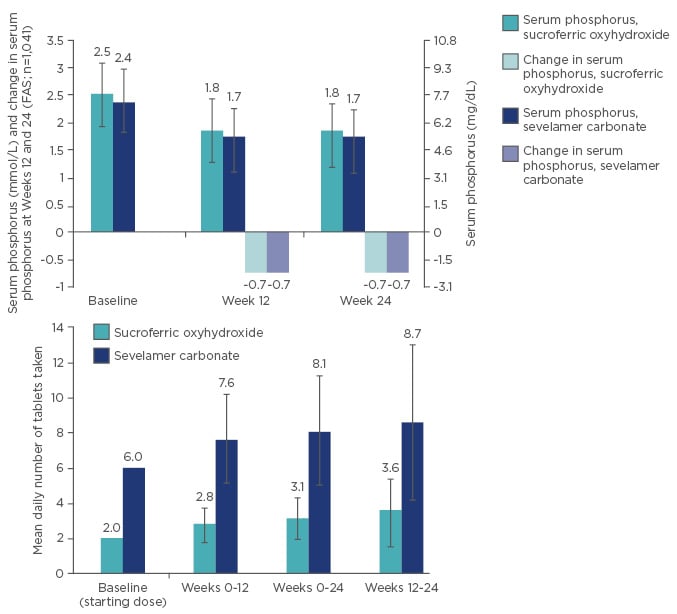
Figure 2: Non-inferiority and lower daily pill burden of sucroferric oxyhydroxide (SFOH) versus sevelamer carbonate.
FAS: full analysis set.
GI disorders were the most frequent treatment-emergent adverse events in both treatment groups, observed in 45.1% of SFOH-treated patients and 33.6% of sevelamer carbonate-treated patients. The incidence of GI-related treatment-emergent adverse events, excluding stool discolouration (due to the iron component), was similar between SFOH (39.0%) and sevelamer carbonate (33.3%) groups. Diarrhoea was reported in 17% of SFOH treated patients during the titration phase of the study but in the maintenance phase diarrhoea had fallen to 5.5% in the SFOH-treated patients compared with 2% in the sevelamer-treated group. Diarrhoea generally presented early in treatment and resolved without treatment change.30 Looking at iron parameters from baseline to Week 24, median serum ferritin concentrations increased in both treatment groups and a statistically significant but not clinically important increase in median transferrin saturation was observed in the SFOH group. These increases occurred early and plateaued on continuing treatment with SFOH, indicating no accumulation of iron. There were also no significant changes in haemoglobin parameters.30 Over the extension period of the study (to 52 weeks), reduction in serum phosphate concentrations was maintained for both treatment groups, while incidence of the most frequent GI disorders fell.32 The extension study has also shown a generally higher adherence with SFOH compared with sevelamer at the pre-defined 70–120% compliance level. Adherence increased over the course of the entire 52 weeks in the SFOH group, possibly due to the lower pill burden in that treatment group over the whole treatment period.31 There was also a 50% lowering in the FGF-23 levels in 1,055 patients in both treatment groups from baseline over 1 year.33
A retrospective database analysis of 693 haemodialysis patients with poorly controlled hyperphosphataemia prescribed SFOH in routine clinical practice was conducted at US Fresenius Medical Care facilities. Observation periods included a baseline period of 3 months before SFOH prescription, and follow-up of 6 months of SFOH prescription. Serum phosphorus at baseline in these patients was 6.9 mg/dL. An increase in the number of patients achieving targets for serum phosphorus was observed after switching from other phosphorus binders to SFOH, this included sevelamer, lanthanum, and calcium acetate. In addition, reduction in the pill burden was observed (8.6 versus 3.4 pills per day).34 The analysis confirmed the efficacy of SFOH as well as the reduced pill burden that may translate into improved adherence in a real-world population.34
In summary, phosphorus homeostasis is lost in CKD. Impaired renal function results in loss of phosphorus excretion, leading to inevitable hyperphosphataemia, a risk factor for CV disease, vascular and other soft-tissue calcification, bone disease, and increased mortality. Management of hyperphosphataemia through dialysis, diet, and phosphate binders is still far from optimal and better treatment options are needed. Several studies have shown that NCCPB appear to attenuate calcification. Adherence to high pill load is a problem in managing phosphate in dialysis patients but SFOH (Velphoro) now presents a new alternative NCCPB therapy as an efficacious option that has a low pill requirement.