Meeting Summary
Secondary hyperparathyroidism (SHPT) can manifest early in chronic kidney disease (CKD) and is associated with significant morbidity and mortality. As such, it is a key feature of CKD that should be assessed and treated from early in the disease. Parathyroid hormone (PTH) levels, greatly increased above reference ranges in SHPT, are controlled via vitamin D receptors; hence, there is an intimate relationship between levels of vitamin D and PTH, as the former suppresses the latter. Clinical studies show that people with CKD may require higher serum 25-hydroxyvitamin D (25[OH]D; calcifediol) target levels than those without CKD to compensate for decreased kidney activation of 25(OH)D to 1,25-dihydroxyvitamin D (1,25[OH]2D; calcitriol). While SHPT can be managed by addressing vitamin D insufficiency, current approaches have limitations in that they either do not reliably and consistently lower PTH levels, as shown with cholecalciferol and ergocalciferol, or they can increase hypercalcaemia risk, as shown with active vitamin D/analogues. Extended-release calcifediol can control SHPT by steadily increasing 25(OH)D levels over time, leading to sustained lower PTH levels with minimal increases in levels of calcium or phosphorus.
Introduction
Chronic kidney disease can result in morbidity and mortality not only due to renal dysfunction itself, but also to associated conditions.1 Declining kidney function can result in poor phosphate excretion, with subsequent increases in serum phosphorus and the bone-derived phosphaturic hormone fibroblast growth factor (FGF-23), along with reduction in vitamin D activation from 25(OH)D (calcifediol) to 1,25(OH)2D (calcitriol).2
Consequences of such renal dysfunction include disruption in bone mineral metabolism, which can lead to vascular calcification and subsequent cardiovascular disease; bone abnormalities, which can increase fracture risk; and parathyroid gland hyperplasia, due to increased parathyroid hormone synthesis and release, known as SHPT.2,3 As such, recognition and treatment of CKD mineral and bone disorder (CKD-MBD) parameters are vital to avoid these potentially life-altering, and sometimes fatal, complications.
Here the author provides an overview of a presentation by Michael Germain, focusing on SHPT. Germain is a transplant nephrologist at Bay State Medical Centre in Springfield, Massachusetts, USA, and a Professor of Medicine at Tufts University School of Medicine in Boston, Massachusetts, USA. This symposium was delivered as part of the ERA-EDTA 58th Congress (Virtual), 5th–8th June 2021.
Vitamin D Catabolism and Deficiency
Vitamin D, from ultraviolet B radiation (as vitamin D3, cholecalciferol) or dietary sources (as vitamin D2, ergocalciferol; or D2), is converted first in the liver to 25(OH)D then in the kidney, parathyroid gland, or other extrarenal tissue such as macrophages, endothelial cells, and the colon, to the active form 1,25(OH)2D.4 This latter conversion, catalysed by the enzyme 1-α-hydroxylase, can be regulated by PTH and FGF-23.4-6 Both of these forms of vitamin D can be catabolised by 24-hydroxylase to inactive forms, respectively 24,25(OH)2D and 1,24,25(OH)3D.
Deficiency in vitamin D is generally defined as serum 25(OH)D levels <20 ng/mL, with insufficiency being 20−29 ng/mL.7 Germain explained how in North-Eastern USA, and presumably other countries in northern latitudes, there is a high incidence of 25(OH)D insufficiency in the general population. This is exemplified by the USA’s National Health and Nutrition Examination Survey (NHANES) III 2001−2004 data that revealed very low levels of 25(OH)D (<12 ng/mL) in 3.7% of the general population, with 22.2% showing levels <20 ng/mL.8
In patients with CKD there is increased 25(OH)D and 1,25(OH)2D catabolism and decreased 1,25(OH)2D production because of increased FGF-23.3-9 This exacerbates the effective deficiency of 1,25(OH)2D compared to healthy individuals, leading to a high prevalence in this population of low serum 25(OH)D and 1,25(OH)2D.4,5,9-11 Such deficiencies have been observed in a study of people with CKD where 57% and 14% of patients at Stage 3 (n=65) had low (10−30 ng/mL) or very low (<10 ng/mL) levels of serum 25(OH)D, respectively, with percentages for Stage 4 patients (n=113) being 58% and 26%, respectively.12 In the CKD Stage 5 dialysis population, vitamin D deficiency aggravates to 47% (low) and 42% (very low).13 As patients with CKD with lower 25(OH)D levels have been shown to have higher plasma PTH; this translates to a potential need for patients with CKD to require higher serum 25(OH)D target levels than a healthy population.5,14-16
Secondary Hyperparathyroidism Development
“SHPT is a very long disease,” reported Germain. “It starts very, very early in CKD, before we even see [a patient] and…it progressively goes up.”
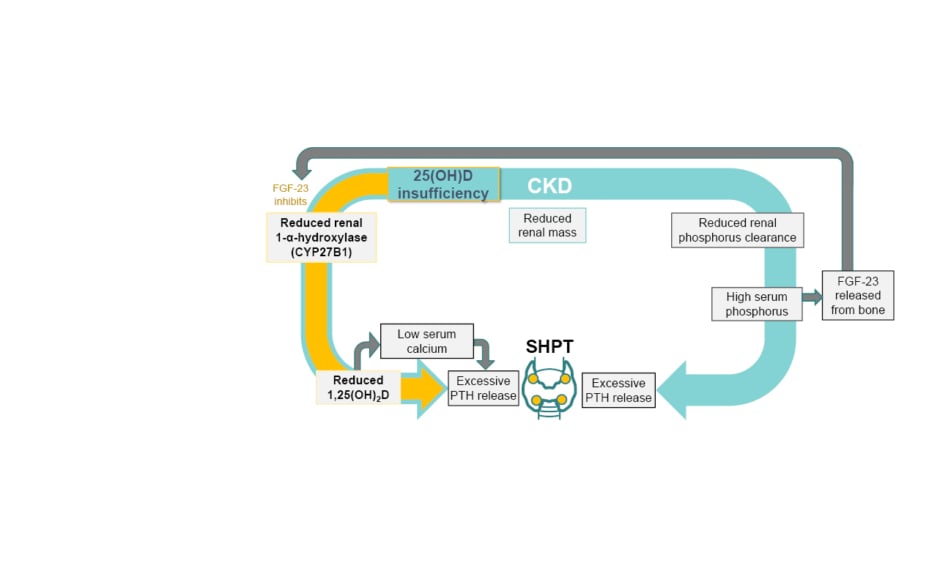
PTH and vitamin D have an intimate relationship whereby PTH stimulates 1,25(OH)2D synthesis, which negatively feeds back to the parathyroid glands to decrease PTH synthesis and release via vitamin D receptors.2 As such, while sufficient levels of 1,25(OH)2D mean PTH suppression, decreased 1,25(OH)2D levels can lead to an increase in PTH release and so to the development of SHPT.2,17 Other factors, such as low serum calcium, increased serum FGF-23, and high serum phosphorus, can also lead to increased PTH synthesis and secretion with diagnosis of SHPT (Figure 1).2,11
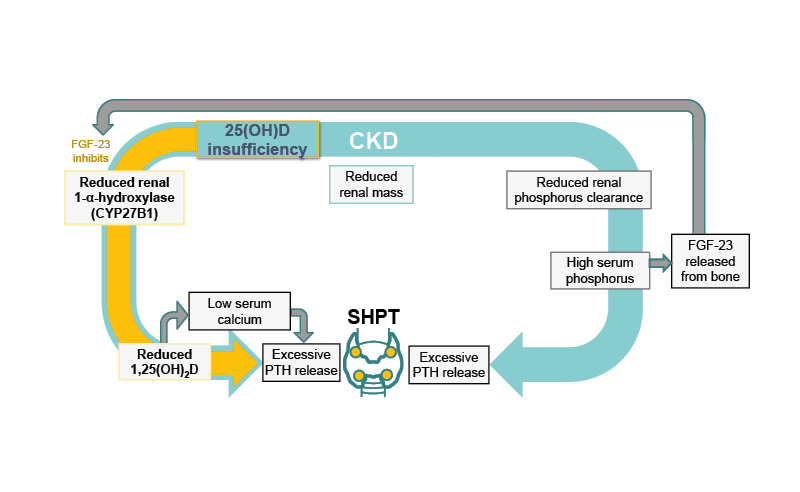
Figure 1: The relationship between low 25-hydroxyvitamin D levels and secondary hyperparathyroidism.2,11
CKD: chronic kidney disease; FGF-23: fibroblast growth factor 23; PTH: parathyroid hormone; SHPT: secondary hyperparathyroidism; 1,25(OH)2D: 1,25-dihydroxyvitamin D; 25(OH)D: 25-hydroxyvitamin D.
As highlighted by Germain, SHPT can start early in CKD and become progressively worse with renal decline.2 For instance, a study11 involving 1,814 patients from 153 centres in the USA showed elevated intact PTH (iPTH; >65 pg/mL), denoting SHPT, in approximately 12% of patients with an estimated glomerular filtration rate (eGFR) of 80 mL/min/1.73m2 (Stage 2), with steady increases occurring as CKD progressed so that by late Stage 4/Stage 5 (eGFR <20 mL/min/1.73m2), almost 90% had elevated iPTH.11 Germain noted that in this study, as in his practice, abnormal calcium (<8.4 mg/dL) and phosphorus (>4.6 mg/dL) levels were rarely observed in patients with Stage 2/3 CKD, but these became more prevalent in Stages 4 and 5, to approximately 20% and 40% of patients, respectively.11
Consequences of Secondary Hyperparathyroidism
Germain highlighted that SHPT is important to recognise and treat early as it is associated with poor outcomes and becomes increasingly difficult to control. For patients on dialysis, SHPT can lead to parathyroid gland unresponsiveness to treatments such as activated vitamin D and calcimimetics, resulting in the need for a parathyroidectomy and subsequent need for control of hypoparathyroidism and potential surgical complications. His patient (Case Study) also demonstrated progressively rising PTH levels, which fits in terms of his association with rising risk for associated mortality, vascular events, and fractures.19
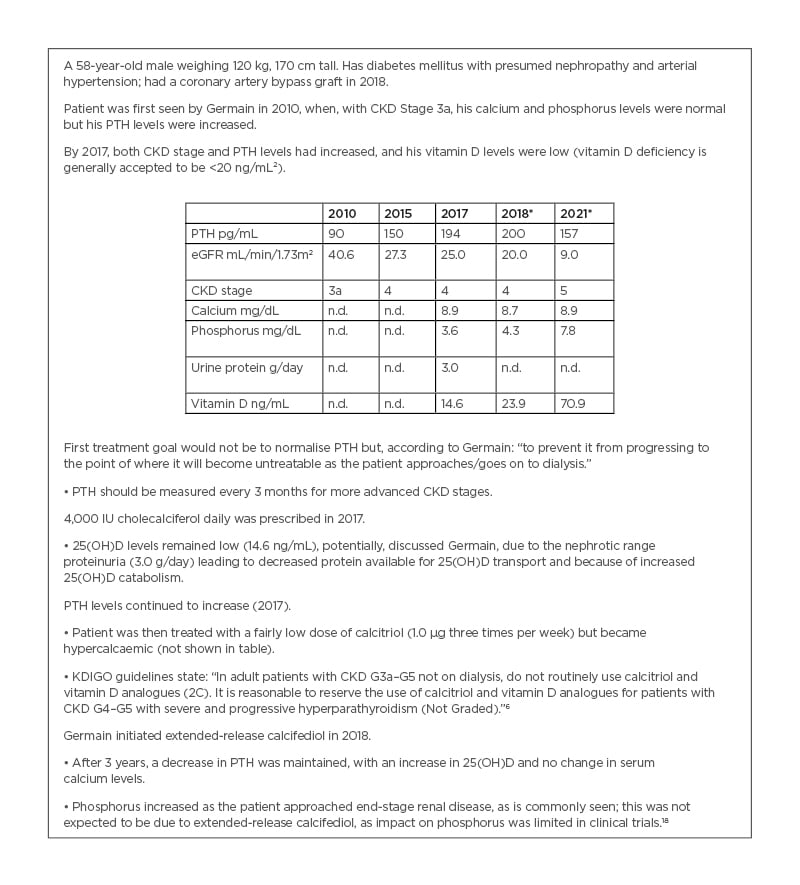
Case study presented by Germain.
A 58-year-old male weighing 120 kg, 170 cm tall. Has diabetes mellitus with presumed nephropathy and arterial hypertension; had a coronary artery bypass graft in 2018.
Patient was first seen by Germain in 2010, when, with CKD Stage 3a, his calcium and phosphorus levels were normal but his PTH levels were increased.
By 2017, both CKD stage and PTH levels had increased, and his vitamin D levels were low (vitamin D deficiency is generally accepted to be <20 ng/mL²).
First treatment goal would not be to normalise PTH but, according to Germain: “to prevent it from progressing to the point of where it will become untreatable as the patient approaches/goes on to dialysis.”
• PTH should be measured every 3 months for more advanced CKD stages.
4,000 IU cholecalciferol daily was prescribed in 2017.
• 25(OH)D levels remained low (14.6 ng/mL), potentially, discussed Germain, due to the nephrotic range proteinuria (3.0 g/day) leading to decreased protein available for 25(OH)D transport and because of increased 25(OH)D catabolism.
PTH levels continued to increase (2017).
• Patient was then treated with a fairly low dose of calcitriol (1.0 µg three times per week) but became hypercalcaemic (not shown in table).
• KDIGO guidelines state: “In adult patients with CKD G3a–G5 not on dialysis, do not routinely use calcitriol and vitamin D analogues (2C). It is reasonable to reserve the use of calcitriol and vitamin D analogues for patients with CKD G4–G5 with severe and progressive hyperparathyroidism (Not Graded).”6
Germain initiated extended-release calcifediol in 2018.
• After 3 years, a decrease in PTH was maintained, with an increase in 25(OH)D and no change in serum calcium levels.
• Phosphorus increased as the patient approached end-stage renal disease, as is commonly seen; this was not expected to be due to extended-release calcifediol, as impact on phosphorus was limited in clinical trials.18
* ERC 30 μg initiated 2018.
CKD: chronic kidney disease; eGFR: estimated glomerular filtration rate; ERC: extended-release calcifediol; n.d.: no data; PTH: parathyroid hormone.
These poor outcomes are illustrated by an observational study that included 5,108 patients with Stages 3−4 CKD, showing that a steady increase in PTH levels was associated with an increase in the 10-year probability of fractures, vascular events, and death.19 In another study,20 involving 2,728 patients with CKD, elevated PTH prior to dialysis was strongly associated with uncontrolled SHPT during dialysis. Regardless of the use of active vitamin D or calcimimetics at this stage, patients who initiated haemodialysis with PTH >600 pg/mL had a 19% higher risk of untreatable SHPT compared to patients with 150−300 pg/mL levels of PTH.20
Germain highlighted some of the consequences of SHPT that he sees in his patients. Active bone disease, which increases the risk of fractures, can be seen in the fingers, with clear radial side and finger tuft bone resorption (Figure 2A). Increased vascular calcifications, shown in the abdominal vessels (Figure 2B), is, discussed Germain, associated with a risk for cardiovascular disease and peripheral vascular disease, as well as potentially preventing a patient from being able to undergo a kidney transplant due to calcified vessels not being amenable to anastomosis onto a transplanted kidney.
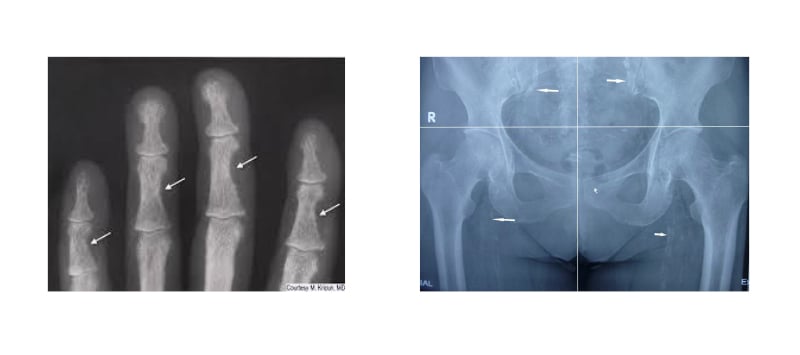
Figure 2: Consequences of secondary hyperparathyroidism.
A) Subperiosteal bone resorption in fingers. B) Vascular calcification of the hip and pelvis.
Treatment for Secondary Hyperparathyroidism
As optimal PTH levels in people with CKD are not known, the Kidney Disease Improving Global Outcomes (KDIGO) clinical practice guidelines for non-dialysis patients with CKD (Stages 3a−5) recommend that in patients where levels of iPTH are progressively rising or are persistently above the upper normal limit, evaluation should be made of modifiable factors, including hyperphosphataemia, hypocalcaemia, high phosphate intake, and vitamin D deficiency.6 They suggest that for those on dialysis, the iPTH range should be maintained between 2−9 times the upper limit of the normal range.6
An ideal treatment for SHPT would increase 25(OH)D levels to physiologic ranges only, decrease PTH levels, and have no impact on levels of calcium, phosphorus, and FGF-23 (Table 1).21
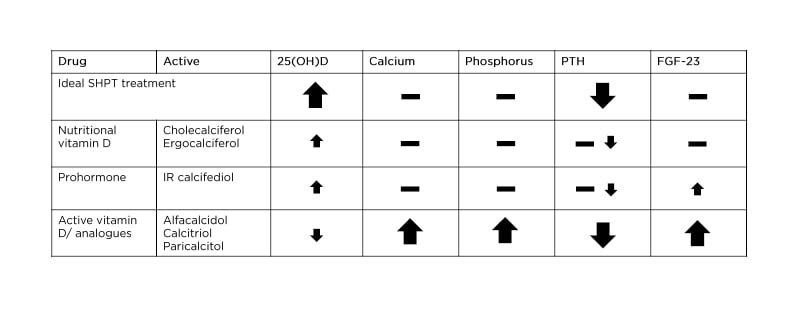
Table 1: Effect of current treatment options on secondary hyperparathyroidism parameters in non-dialysis/dialysis patients with chronic kidney disease.21,22,23-26
FGF-23: fibroblast growth factor 23; IR: immediate-release; PTH: parathyroid hormone; SHPT: secondary hyperparathyroidism; 25(OH)D: 25-hydroxyvitamin D.
In people with CKD (non-dialysis or dialysis), 25(OH)D supplementation with cholecalciferol or ergocalciferol is the initial recommendation for 25(OH)D deficiency, with vitamin D prohormone, as immediate-release (IR) calcifediol, also considered.6,21,22 However, these treatments only moderately increase 25(OH)D levels and have a non-reliable and non-consistent effect on PTH, with IR calcifediol slightly increasing levels of FGF-23, which can lead to breakdown of 25(OH)D and 1,25(OH)2D3 and reduced generation of 1,25(OH)2D3 (Table 1).21,27,28
Active vitamin D (1,25[OH]2D3) or vitamin D analogues, such as alfacalcidol, calcitriol, or paricalcitol, may be considered next for both non-dialysis and dialysis patients (Table 1).21,24-26 However, while vitamin D analogues can suppress PTH more strongly, they can also result in a large rise in calcium, phosphorus, and FGF-23 levels, all without a substantial beneficial effect on serum 25(OH)D.21,29
Extended-Release Calcifediol
A study in rats showed that administration of IR calcifediol led to high serum levels of both 25(OH)D, with an immediate spike following injection, and 1,25(OH)2D, with a spike a few hours later.23 Of note, levels of 25(OH)D remained very high for at least 25 hours post-injection and, notably, the amount of detectable 1,25(OH)2D dropped to near-baseline levels by 25 hours.
These findings in an animal study fit into the postulate that pharmacological surges of serum 25(OH)D may trigger negative feedback of endogenous vitamin D signalling, inducing catabolism.5 Indeed, this was shown in a clinical study where the same 25(OH)D initial spike seen in rats was shown in participants who received a 448 mg bolus of IR calcifediol, with levels remaining high for at least 4 days.
Of interest, 25(OH)D catabolism, as illustrated by 24,25(OH)2D levels in this study, increased over a period of 12 days then remained high (around 40 ng/mL) until at least Day 40 post-injection. This was postulated to be due to the 25(OH)D spike from IR calcifediol increasing levels of the hormone responsible for 25(OH)D breakdown, 24-hydroxylase, and of FGF-23 (Figure 1), as found in the rat study.23
A more recent development for SHPT treatment is extended-release (over 12 hours) calcifediol (ERC; European Union [EU] term: prolonged release calcifediol).21,23 In contrast to the IR formulation, in the animal experiment, while administration of ERC still led to some increases of 25(OH)D levels at 5 hours post-dose, this was to within physiologic levels, which gradually fell over the next 20 hours. There was also a much gentler rise in 1,25(OH)2D, that, unlike with IR calcifediol, led to increasing levels of this active form of vitamin D over time (up to 25 hours post-dose). These were far lower levels than with the IR formulation and kept within the physiologic range. In the human study, a single dose of extended-release calcifediol (450 or 900 mg) did not lead to a spike in 25(OH)D but achieved a steady-state level over 4 days. There was decreased 25(OH)D catabolism, shown by significantly lower 24,25(OH)2D levels compared to IR calcifediol over the 40 days post-administration.23 This, suggested Germain, meant that extended-release calcifediol could “normalise the homeostatic response to vitamin D by PTH.”
To examine the impact of ERC on not only 25(OH)D but also PTH, calcium, and phosphorus levels, two Phase III clinical trials (Study A and Study B) of ERC were carried out, including a total of 429 adults with non-dialysis CKD Stage 3−4, SHPT (iPTH: 85–499 pg/mL), vitamin D insufficiency (25[OH]D: 10–29 ng/mL), calcium levels 8.4–9.7 mg/dL, and phosphorus levels 2.0–4.9 mg/dL.18
Participants were stratified by CKD stage and received either 30 μg ERC daily or placebo for 12 weeks in a 2:1 ratio. For the following 14 weeks, the ERC group either remained on 30 μg ERC daily or the dose was increased to 60 μg if PTH >70 pg/mL, 25(OH)D ≤65 ng/mL, and serum calcium <9.8 mg/dL. In an open-label extension, the placebo group was switched to 30 μg ERC daily for 12 weeks, with those taking ERC remaining on the same dose. After 12 weeks, any participant with PTH >70 pg/mL and serum calcium <9.8 mg/dL were switched to 60 μg ERC daily for 14 weeks.18
The primary endpoint to this study, a ≥30% decrease in iPTH from baseline, was met by both studies (Figure 3A), with 33% and 34% of patients in Studies A and B, respectively, achieving this at Week 26 with ERC, compared with 8% and 7% of placebo patients, respectively.18
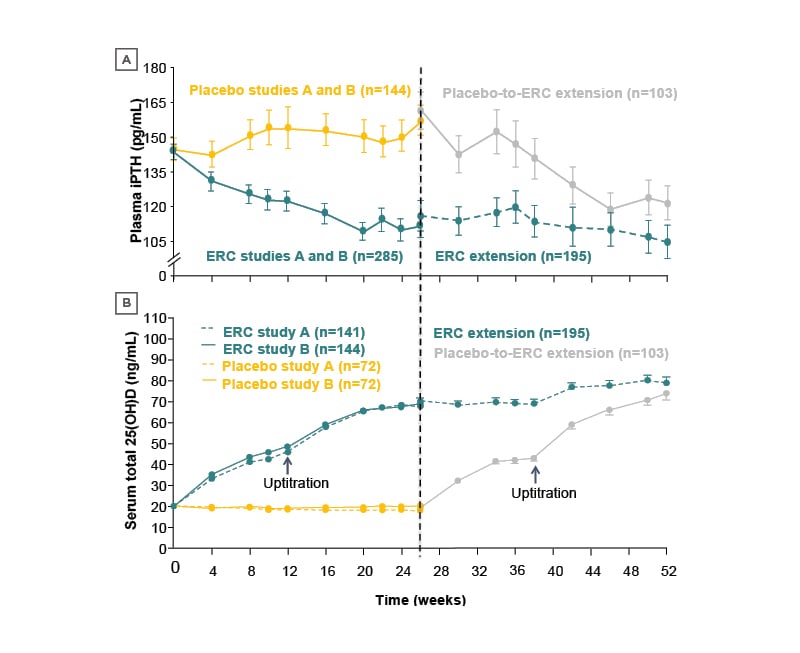
Figure 3: Mean standard error change over time in A) plasma-intact parathyroid hormone and B) serum total 25-hydroxyvitamin D (per protocol population).18
Note: Error bars are omitted for clarity in the first portion of the study.
ERC: extended-release calcifediol; iPTH: intact parathyroid hormone; 25(OH)D: 25-hydroxyvitamin D.
This decrease in iPTH was accompanied by an increase in 25(OH)D levels to 30−100 ng/mL8 in patients treated with ERC; this was also achieved when placebo participants were switched to ERC (Figure 3B).18 At Weeks 26 and 52, 25(OH)D levels were 50 and 59 ng/mL, respectively, for patients on the 30 μg dose of ERC, and 70 and 82 ng/mL, respectively, for patients on the 60 μg dose of ERC.30
Mean serum phosphorus levels were slightly greater in the ERC group, with a mean change from baseline of 0.2 mg/dL (standard error [SE]: 0.03) versus 0.1 mg/dL (SE: 0.04) in the placebo groups. Near-identical differences (0.2 mg/dL, SE: 0.02; versus 0.1 mg/dL, SE: 0.03) were shown for rises in serum calcium (p<0.0001). In the majority of patients, these increases had no clinical implications, with only one patient in the ERC group meeting the definition of hyperphosphataemia (two consecutive serum phosphorus values >5.5 mg/dL deemed study-related) and six participants in the ERC group who required dose reductions for hypercalcaemia (two consecutive serum calcium values >10.3 mg/dL). There were no significant differences between placebo and ERC or between ERC doses for serum FGF-23.18,21
Another finding of these studies (Figure 4) was that those with the highest 25(OH)D levels (mean: 92.5 ng/mL) had the greatest PTH suppression at Weeks 20−26 (to a mean of 97 pg/mL) and, conversely, those with the lowest 25(OH)D levels (mean: 13.9 ng/mL) had the least PTH suppression at Week 26 (mean: 166 pg/mL).14 This, advised Germain, means that: “If you follow your vitamin D level, you can have a good indication of the efficacy you’re going to get with ERC.”
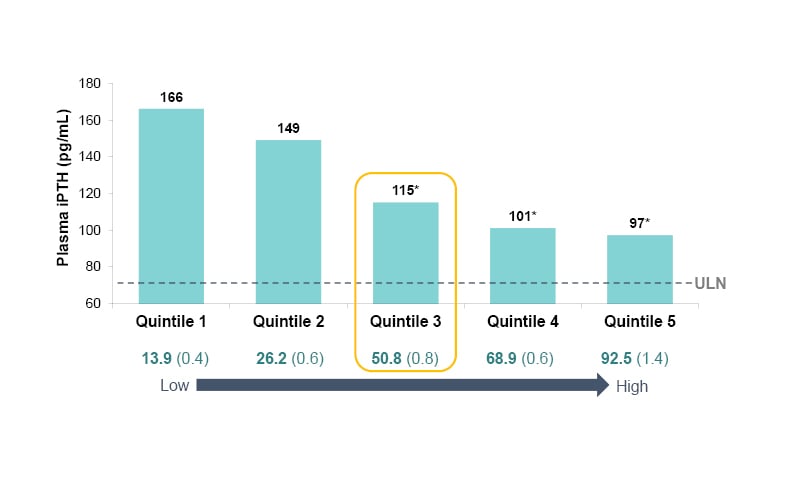
Figure 4: Plasma parathyroid hormone at Weeks 20−26 of treatment with extended-release calcifediol (30 or 60 μg) or placebo, as a function of post-treatment 25-hydroxyvitamin D quintiles (per-protocol population).14
*p<0.0001 versus quintile 1.
ERC: extended-release calcifediol; iPTH: intact parathyroid hormone; ULN: upper limit of normal; 25(OH)D: 25-hydroxyvitamin D.
While clinical trials can reveal a lot of information about a drug, study participant limitations mean findings may not necessarily translate to a more general population. In a real-world setting, a retrospective analysis31 was carried out of 174 patients with Stage 3−4 CKD, SHPT, and 25(OH)D insufficiency, of whom 173 received 30 μg daily ERC and one received 60 µg daily. This analysis found that after 12 months of ERC treatment, 70.1% of 122 analysed at this timepoint achieved 25(OH)D target levels of ≥30 ng/mL, with 40.2% of 70 patients analysed at this timepoint showing ≥30% reduction in PTH. There was little difference in calcium and phosphorus levels with continued ERC dosing in this cohort when analysed at a mean 27.8 and 28.8 weeks of follow-up, respectively.31
Conclusion
As SHPT manifests early in CKD and is associated with significant morbidity and mortality, it is a key feature of CKD that should be assessed and treated from the early stages of the disease.2 Clinical studies show that people with CKD may require higher serum 25(OH)D target levels (>50 ng/mL) than those without CKD to achieve PTH reduction.5,14,15 While SHPT can be managed by addressing vitamin D insufficiency, current approaches have limitations in that they either do not reliably and consistently lower PTH levels, as shown with cholecalciferol and ergocalciferol,22 or they can increase hypercalcaemia risk, as shown with active vitamin D/analogues.32 ERC can control SHPT by steadily increasing 25(OH)D levels over time, thus not inducing excess 25(OH)D catabolism, leading to lower PTH levels with minimal clinically relevant increases in calcium or phosphorus.6,16,31