Abstract
The rapid increase in the global incidence of obesity presents an ever-expanding set of medical, social, and economic challenges. Obesity is a complex disease, with the emphasis of current research aimed at unravelling its link with a range of health conditions.
The chronic inflammatory state created by obesity is frequently postulated as the driving force behind its pathophysiological consequences in a number of health conditions. Specifically, there is expanding evidence linking obesity to the development of a number of central and peripheral nervous system disorders.
This review evaluates established and emerging evidence for the link between adiposity and a range of neurological phenomena. Knowledge of the pathological mechanisms of obesity may prove useful in determining the most effective treatments of obesity-induced neurological disorders.
Key Points
1. Obesity is a complex disease linked to a range of health conditions, including neurological conditions affecting both the CNS and PNS. The recent rapid increase in its global incidence presents expanding medical, social, and economic challenges.2. The chronic inflammatory state created by obesity is frequently postulated as the driving force behind pathophysiological processes in a range of neurological phenomena.
3. The increasing prevalence of obesity necessitates further investigation into the pathways behind potential nervous system damage caused by obesity, and highlights the need for comparing already established dietary, physical, pharmacological, and surgical weight loss interventions.
INTRODUCTION
Obesity is defined by the National Institute of Health and Care Excellence (NICE) as a body mass index (BMI) greater than 30 kg/m². Recognised as a chronic and progressive disease, obesity is linked with a number of comorbidities that have a detrimental impact on health. The current obesity epidemic is a major public health problem across the world. In 2016, approximately 13% of the world’s adult population were obese, with the incidence of obesity almost tripling between 1975 and 2016.1 It is estimated that by 2050, 60% of males and 50% of females will be obese.2 A considerable burden on health services is anticipated, due to the increasing incidence of Type 2 diabetes, hypertension, cardiovascular disease, stroke, osteoarthritis, sleep apnoea, and certain cancers, amongst other conditions linked to obesity. The social and economic impacts of obesity are equally recognised.2
Expanding evidence now associates obesity as a risk factor for an array of neurological disorders.3 Obesity-driven dysfunction drives central nerous system (CNS) and peripheral nervous system (PNS) damage via complex mechanisms.4 It is well known that visceral adiposity has metabolic and endocrine implications, impacting on various organs, including the liver, pancreas, and vasculature, resulting in hyperinsulinemia, hyperglycaemia, dyslipidaemia, and hypertension.5 This review describes the link between obesity and disorders of the CNS and PNS. The authors outline the recent research in this field, whilst examining how lifestyle and surgical interventions can reduce the incidence of neurological conditions, where obesity is a known risk factor.
SEARCH STRATEGY
A literature search was performed using Medline and PubMed for papers published between 2000 and 2021. The following keywords were used in various combinations: ‘obesity’, ‘BMI’, ‘high fat diet’, ‘neurological disorders’, ‘inflammation’, ‘Alzheimer’s disease’, ‘autonomic nervous system’, ‘peripheral nervous system’, ‘bariatric surgery’, ‘dyslipidaemia’, and ‘adiposity’. Studies that were not written in English were excluded, as well as those that did not contain more than one of the keywords in the title. Furthermore, studies with small sample sizes and those with abstracts not relevant to the search were excluded.
PATHOPHYSIOLOGY OF OBESITY AND NEUROLOGICAL DYSFUNCTION
Adipose tissue regulates whole body homeostasis and is a key organ in the modulation of energy storage. Its involvement in metabolic and immune processes is thought to play a leading role in obesity-induced complications. A high-fat diet and reduced energy expenditure yields an overall positive energy balance. Excess lipid accumulation in adipose tissue and increasing visceral adipose mass prompts hypertrophy and hyperplasia of adipocytes, triggering their dysfunction.6 Metabolic inflammation occurs in hypertrophied adipocytes due to the activation of stress signalling pathways. This results in the release of free fatty acids (FFA), proinflammatory cytokines, and altered adipokine levels. A hypoxic state in adipocytes is created, triggering the infiltration of macrophages, which further contributes to the release of cytokines.7,8
Obesity is characterised by a chronic low-grade inflammation that is considered the driving force behind obesity-induced neurological disorders.4 The pathophysiology behind neuronal damage is complex and multifactorial with multiple pathways involved, including lipotoxicity, insulin resistance, and oxidative stress (Figure 1). Alterations in these signalling pathways result in damage to cells, and eventually in neuronal loss. Below are four postulated mechanisms.
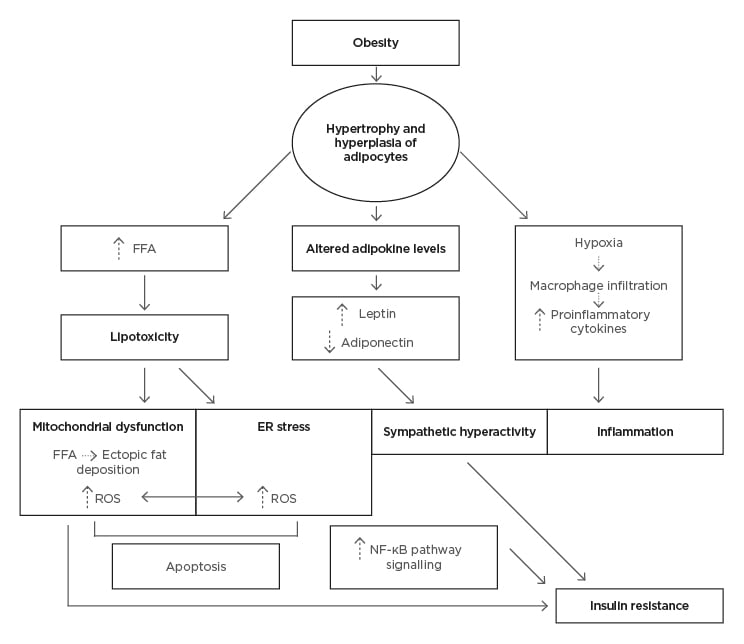
Figure 1: Pathological mechanisms of obesity induced dysfunction in the central nervous system
ER: endoplasmic reticulum; FFA: free fatty acid; ROS: reactive oxygen species.
Neuroinflammation
Obesity results in the release of proinflammatory cytokines such as TNFα, IL-6, and IL-1β, which trigger neuroinflammation and result in subtle changes in brain morphology.8 Such neuroinflammation has been associated with lower cortical grey and white matter volumes, and the onset of cognitive impairment.9,10 Thaler et al.11 put forward hypothalamic inflammation as the first stage of neuroinflammation in the CNS, as proinflammatory cytokines and FFAs increase the permeability of the blood–brain barrier.
With regard to the PNS, the blood–nerve barrier, dorsal root ganglion, and free nerve endings are susceptible to injury via inflammatory mechanisms in the context of normoglycaemia and hyperglycaemia, leading to polyneuropathy in the population with obesity.12 It is the chronic inflammation caused by obesity that is injurious to peripheral nerves and underlies the pathogenesis of neurological disorders associated with obesity.
Adipokine Levels
Obesity induces changes in circulating adipokine levels from adipose tissue.13 Adipokines play a homeostatic role as intermediaries between metabolic and immune function. Mice with obesity demonstrate decreased levels of adiponectin, a cytokine released from white adipose tissue that is able to cross the blood–brain barrier to exert anti-inflammatory effects.14 The suppression of the adiponectin receptor 1 contributes to insulin resistance in the brain. A long-term, high-fat diet can thus impair insulin signalling, which is important for synaptic plasticity and glucose homeostasis, resulting in the death of neuronal cells and exacerbating neurodegeneration. Animal studies have substantiated the association between insulin resistance, cognitive impairment, and neurodegenerative diseases.14,15 Mice deficient in adiponectin present with reduced spatial memory and decreased synaptic protein levels.15
In contrast, increased levels of leptin, visfatin, and resistin are found in patients with obesity. Both visfatin and resistin have been shown to contribute to insulin resistance.16 Obesity decreases the responsiveness to leptin, resulting in a leptin-resistant state, which contributes to synaptic loss and consequently neurodegeneration.17 Leptin receptors in the ventromedial hypothalamic nucleus are desensitised as a consequence of persistently elevated leptin levels, leading to dysregulation of leptin signalling pathways. A vicious circle arises as leptin resistance causes a compensatory increase in leptin release, which further worsens obesity due to the effects of increased sympathetic activity, and suppression of leptin’s anorexigenic function.6 There is still a lack of knowledge on the exact pathogenic mechanisms that adipokines play in obesity. Many mice studies provide an enhanced understanding, with Qatanani et al.16 showing the role of adipokines being assignable to human disease.
Free Fatty Acids
Obesity increases basal lipolysis and the generation of FFAs in adipose tissue. Increased cellular uptake of FFAs causes lipid overload, thereby resulting in lipotoxic effects on nerve cells. The consequences of this include promotion of insulin resistance via ectopic fat deposition in the pancreas and muscles, as well as dyslipidaemia via FFA-induced liver dysfunction.7 These effects lead to metabolic syndrome and are responsible for the increased risk of stroke in the population with obesity.18
Mitochondrial Dysfunction and Endoplasmic Reticulum Stress
Mitochondrial dysfunction is a cause and consequence of lipotoxicity and inflammation. Failure of synaptic remodelling has damaging effects on cells, leading to neurodegeneration and, thus, brain atrophy.7 The lipotoxic state observed in obesity creates an excess of FFAs, which alter the structure of mitochondria, leading to increased mitochondrial depolarisation. Consequently, impaired oxidative phosphorylation causes an increase in reactive oxygen species and decreased adenosine triphosphate production, triggering apoptosis of neurons.19
Moreover, increased FFAs in peripheral nerves contribute to endoplasmic reticulum (ER) stress.20 The ER and mitochondria function together to maintain cellular homeostasis. FFA-induced dysfunction of the ER and mitochondria leads to dysregulated calcium signalling between the two structures, promoting a proapoptotic state, resulting in nerve damage.
CENTRAL NERVOUS SYSTEM
An increased risk of cognitive decline is present in individuals with obesity.9,21 Meta-analyses have exhibited a convincing link between obesity and dementias, including Alzheimer’s disease and vascular dementias.22,23
Studies indicate that the consumption of a high-fat diet leads to widespread oxidative damage24 that is involved in the pathology of neurodegenerative diseases. As demonstrated in mice studies, a high-cholesterol diet can increase amyloid-β protein deposition.25 These changes are reinforced by an insulin-resistant state, which incites Tau hyperphosphorylation and the accumulation of amyloid-β protein.26 These changes are accepted as pathognomonic of Alzheimer’s.
Other theories propose a correlation between obesity and reduced dopaminergic response.27 There is a perceived vicious cycle between insulin resistance and mitochondrial dysfunction, which prompts increased reactive oxygen species levels and neuronal loss. As demonstrated in the substantia nigra, insulin resistance is understood to promote a loss of dopaminergic neurons and augment the expression and signalling of α-synuclein protein, which are hallmarks of Parkinsonian pathology.28,29 Also related to altered function of dopamine in the CNS is the link between obesity and restless legs syndrome.30 A meta-analysis reveals that it is more prevalent amongst individuals with obesity.31
EPILEPSY
It is postulated that epilepsy is related to metabolic dysfunction. One study by Daniels et al.32 has demonstrated obesity to be more prevalent in children with diagnosed epilepsy prior to treatment, compared with controls. Although the relationship between the two is not clear, it suggests that obesity could induce changes in the CNS, leading to increased seizure susceptibility.33 Furthermore, it is widely hypothesised that altered hormone levels lead to altered seizure threshold. Adiponectin-deficient mice fed a high-fat diet demonstrate increased seizure activity, suggesting a protective role against seizures for adiponectin, which is known to be downregulated in obesity.33
NARCOLEPSY AND CATAPLEX
There is a significant association between the chronic sleep disorder narcolepsy and higher BMIs.34 Inocente et al.35 found that 50% of children diagnosed with narcolepsy were obese. An explanation for this may lie in the deficiency of hypocretin, a neurochemical involved in the regulation of the sleep–wake cycle that is proposed to impact on energy homeostasis due to its other roles in body adiposity regulation.34
MIGRAINE
The relationship between migraine and obesity has been extensively researched. A meta-analysis demonstrates increased prevalence of migraine in individuals with BMI within the obese range, with age and sex as important variables within this relationship.36 A population-based study has shown that increased total body and abdominal obesity is associated with higher prevalence of migraine.37 It is suggested that proinflammatory cytokines and altered adipokine levels cause neurogenic inflammation, leading to migraine.
IDIOPATHIC INTRACRANIAL HYPERTENSION
The link between a high BMI and idiopathic intracranial hypertension (IIH) is widely recognised, with weight loss considered the only cure for this condition.38 The exact mechanism behind obesity leading to raised intracranial pressure is unclear, but is related to hormonal change. A lesser-known theory suggests that increased visceral adiposity leads to a rise in intrathoracic and central venous pressure, causing a decrease in intracranial blood flow, resulting in IIH.39
RELATIONSHIP WITH AUTOIMMUNE DISEASES
Evidence suggests a correlation between obesity in adolescence and a higher risk of developing multiple sclerosis (MS).40 This study demonstrates adolescents with a BMI >27 kg/m2 having a higher risk of developing MS, compared with those of a BMI 18.5–21.0 kg/m2. Also, there is a correlation between the MS risk gene HLA-DRB1*15 and adolescent obesity. There are many potential mechanisms for this relationship, with leptin playing a leading role in linking metabolic changes with immune dysfunction. It is proposed that leptin favours the induction of an inflammatory state, which underlies the pathology of MS, by inducing Th1 responses and decreasing the activity of regulatory T cells, contributing to the development of autoimmune diseases.41
Dyslipidaemia can also be linked to the onset of autoimmune disease. A study by Wu et al.42 demonstrates that higher levels of triglycerides may worsen the prognosis of the rare condition neuromyelitis optica. Increased levels of cytokines and chemokines have been identified in the pathogenesis of both MS and neuromyelitis optica.43 Therefore, it is plausible that the inflammatory state created by obesity could play a pathophysiological role and worsen patient outcomes in these neuro-inflammatory conditions.
NEUROPSYCHIATRIC DISORDERS
A rodent study suggests a high-fat diet could induce core symptoms of depression, such as anxiety and anhedonia.27 In this case, diet duration played a key role in the changes propagated by obesity. Further evidence indicates obesity is associated with neuropsychiatric diseases such as bipolar disorder and schizophrenia, in part due to obesity-induced mitochondrial dysfunction and inflammation.4,21,44
PERIPHERAL NERVOUS SYSTEM
The PNS is composed of autonomic and somatic divisions that have also been demonstrated to be vulnerable to the pathophysiological consequences of obesity. The somatic nervous system functions to control the body’s voluntary movements. Obesity can trigger damage to sensory and motor nerves, as illustrated in Figure 2.
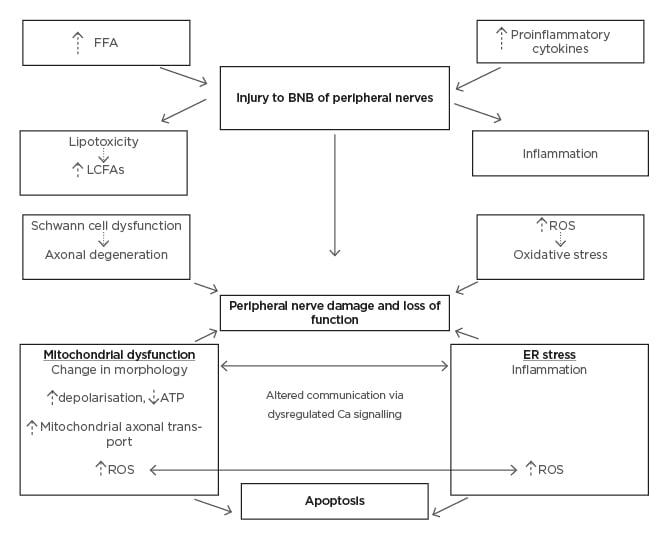
Figure 2: Pathological mechanisms of obesity induced dysfunction in the peripheral nervous system.
ATP: adenosine 5′-triphosphate; BNB: blood–nerve barrier; Ca: calcium; ER: endoplasmic reticulum; FFA: free fatty acid; LCFA: long-chain fatty acid; ROS: reactive oxygen species.
A sensory nerve consists of a bundle of neurons surrounded by supportive connective tissue. Neurons are comprised of axons, dendrites, and cell bodies in the dorsal root ganglion. Cell bodies are dependent on mitochondria for energy to function, such that damage to mitochondria can heavily impact on normal nerve function. This is one postulated mechanism leading to polyneuropathy.19 It is well documented that diabetes is an important risk factor for polyneuropathy, typically causing sensory and motor deficits in a distal to proximal manner.19 However, emerging data now elucidates obesity to be one of the principal metabolic triggers for polyneuropathy, independent of diabetes and pre-diabetes. A cross-sectional study shows a significant association between waist circumference and neuropathy.12 Chemokines C-C motif ligand 7 and C-X-C motif chemokine ligand 10 are known to be released in a distal symmetric polyneuropathy.45 High levels of these chemokines can result in nerve structure damage and are thought to contribute to pain in polyneuropathy.
A prospective analysis shows a correlation between higher BMI and waist circumference, and an increased risk of Guillain–Barré syndrome.46 Likewise, a positive correlation between high BMI and Bell’s palsy has been demonstrated in a case control study.47 A slower recovery rate in patients with obesity and Bell’s palsy has been demonstrated.48 These relationships could be, in part, plausibly explained by the pre-disposing inflammatory state created by obesity.
Obesity is additionally associated with an increased incidence of mononeuropathies. A relationship is demonstrated between raised BMI and carpal tunnel syndrome, as well as meralgia paresthetica.49-51
AUTONOMIC NERVOUS SYSTEM
The autonomic nervous system is divided into sympathetic and parasympathetic divisions, and plays a role in homeostatic energy balance by regulating food intake and energy expenditure. Studies demonstrate that the presence of autonomic nervous system dysfunction in individuals with obesity.52 Obesity is perceived to provoke a selective hyperactivity of the sympathetic divisions via an impaired baroreceptor reflex and increased leptin and insulin levels.53 Both leptin and insulin bind to receptors in the CNS, which are selective for the regulation of sympathetic tone. This overactivity causes increased stimulation of β-adrenergic receptors in neuro-adipose junctions, promoting lipolysis and increased FFA levels.54 Greater sympathetic stimulation of skeletal muscles also precipitates insulin resistance, due to changes in glucose metabolism (Figure 3).52
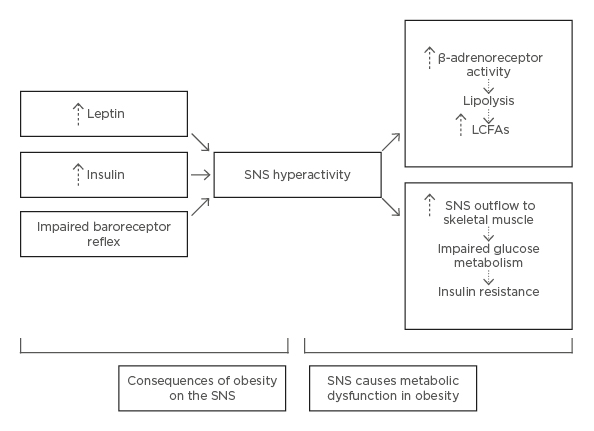
Figure 3: Pathological mechanisms of obesity induced dysfunction in the sympathetic nervous system.
LCFA: long-chain fatty acids; SNS: sympathetic nervous system.
A two-way relationship is observed between obesity and autonomic nervous system dysfunction. Obesity can trigger changes to autonomic nervous system signalling, resulting in metabolic and cardiovascular consequences; conversely, increased sympathetic stimulation can propagate metabolic dysfunction. Studies demonstrate that weight loss in individuals with obesity causes a decrease in sympathetic activity in muscles, further supporting this relationship and the potential for therapy that targets the sympathetic nervous system to improve metabolic profile.55
NEUROLOGICAL DISORDERS ASSOCIATED WITH OBESITY
Overall, from the extensive data analysed, it is evident that obesity has a pathological role in CNS and PNS dysfunction. There are numerous neurological disorders known to be associated with obesity, which are summarised in Table 1.

Table 1: List of neurological disorders associated with obesity.
TARGETING OBESITY IN NEUROLOGICAL DISORDERS
It is widely recognised that targeting obesity can generate considerable health benefits in an individual, attributable to the physiological benefits that arise, such as improved glucose homeostasis, lipid levels, and vascular function. This leads to enhancements in the overall systemic and metabolic health of an individual. Given the multiple chronic diseases correlated with obesity, its prevention can be seen as essential in minimising their risk.
Improved cognitive performance is demonstrated when obesity is targeted.56 A combination of dietary and exercise mediations are preferred means in tackling obesity and its complications. A 2-year randomised controlled trial has demonstrated that increased exercise, cognitive training, and changes to diet are successful in preventing cognitive decline.57
Nonetheless, compliance with these lifestyle changes can sometimes be difficult, and this is when pharmacological and surgical methods are sometimes required. Bariatric surgery has proven to be an extremely effective treatment for patients with morbid obesity. Various studies demonstrate improvements in neurological function following this procedure.58 One study has illustrated decreased inflammation and amyloid precursor protein expression following bariatric surgery.59 These are hallmark changes in Alzheimer’s disease and indicate that weight loss after such interventions could have beneficial effects on brain structures, leading to improved cognitive function. Furthermore, IIH in a subject with obesity is an absolute indication for bariatric surgery, which is effective in producing significant improvements in symptoms, or complete remission.60
Bariatric surgery is highly successful in treating weight loss, but carries with it multiple complications.59 Many complications, such as polyneuropathy, are driven by the after-effects of nutrient deficiency. Adequate vitamin and micronutrient supplementation is required post-operatively to prevent such neurological complications.
CONCLUSION
Obesity has been demonstrated as a risk factor for a range of neurological disorders, affecting both the CNS and PNS. A complex set of mechanisms is shown to be involved in the development of neurological dysfunction. The inflammatory state created by obesity is the driving force behind the pathology, and other mechanisms of insulin resistance, lipotoxicity, mitochondrial dysfunction, ER stress, and sympathetic hyperactivity all interact to provoke further damage.
With the prevalence of obesity ever-increasing, further research is necessary in determining the pathways of potential nervous system damage, as well as comparing the already established interventions of dietary, physical, pharmacological, and surgical weight loss in promoting the best possible health outcomes and reducing the incidence of neurological disorders.