Abstract
Amyotrophic lateral sclerosis is a devastating neurodegenerative condition with few effective treatments. Current research is gathering momentum into the underlying pathology of this condition and how components of these pathological mechanisms affect individuals differently, leading to the broad manifestations encountered in clinical practice. We are moving away from considering this condition as merely an anterior horn cell disorder into a framework of a multisystem neurodegenerative condition in which early cortical hyperexcitability is key. The deposition of TAR DNA-binding protein 43 is also a relevant finding given the overlap with frontotemporal dysfunction. New techniques have been developed to provide a more accurate diagnosis, earlier in the disease course. This goes beyond the traditional nerve conduction studies and needle electromyography, to cortical excitability studies using transcranial magnetic stimulation, and the use of ultrasound. These ancillary tests are proposed for consideration of future diagnostic paradigms. As we learn more about this disease, future treatments need to ensure efficacy, safety, and a suitable target population to improve outcomes for these patients. In this time of active research into this condition, this paper highlights some of the areas of controversy to induce discussion surrounding these topics.
INTRODUCTION
Motor neurone disease (MND) encompasses a group of disorders affecting the anterior horn cells and includes the most common form, termed amyotrophic lateral sclerosis (ALS). The inexorable decline in function leading to death for these conditions, in often otherwise healthy people, has emphasised the unmet need for better diagnostic and treatment paradigms. With awareness of these conditions increasing in the public consciousness, and increasing research funding, this is an excellent opportunity to improve outcomes for these patients.
While previously considered a neuromuscular disorder of the anterior horn cells, greater research in recent years into brain cortical dysfunction, and also identification of deposition of TAR DNA-binding protein 43 (TDP-43) in the brain, has altered the perception of this condition into the category of multisystem neurodegenerative disease. On one end of the spectrum lies predominant anterior horn cell damage and lower motor neuron (LMN) signs. On the other end is cortical TDP-43 deposition leading to behavioural-variant frontotemporal dementia (FTD).
In this review, recent advances in the understanding of the diagnosis and treatment of this condition are discussed. This information is posed as a series of ‘controversial statements’ for which evidence may be conflicting. While the authors have no claim to answer these questions, they hope this will encourage readers to question their own diagnostic and therapeutic preconceptions and generate increased consideration of the underlying issues.
CLASSIFICATION
Amyotrophic Lateral Sclerosis is the Same Disease as Progressive Muscular Atrophy and Primary Lateral Sclerosis
MND encompasses a family of disorders affecting the anterior horn cells, of which ALS is the most common form, demonstrating both upper motor neuron (UMN) and LMN features. Other phenotypes of the disease such as primary lateral sclerosis (PLS) and primary muscular atrophy (PMA) are less common, featuring UMN and LMN deficits, respectively. Even within ALS there is significant variation in the anatomical location and progression of the disease.1 Within ALS, patients may have an UMN- or LMN-predominant symptomatology. There are other phenotypic variants such as the flail arm or flail leg syndrome, and rarer variants such as spinal or diaphragm onset.2,3 From the point of origin, different patterns of disease spread have been characterised.1 Bulbar-onset disease accounts for approximately one-third of ALS cases, and carries a poorer prognosis. Specific genetic changes may influence phenotype and prognosis, but there is significant pleiotropy whereby a mutation may cause a variety of phenotypes, even within the same family.4 Some have incorporated clinical, phenotypic, and genetic factors into a prognostic model.5 A clear indication of prognosis is helpful for patients.
In the absence of a distinguishing biomarker, there has been controversy as to what extent ALS, PMA, and PLS should be defined as a single entity, or to classify them as separate conditions.6 Often with time, other UMN or LMN features become apparent, and the diagnosis is revised to ALS.7,8 In the authors’ opinion, these conditions are likely to be the same pathophysiological condition, albeit with differing location of predominant pathology (and different emphasis on pathological processes) leading to the spectrum of symptoms that they consider in PMA, ALS, and PLS. Some have proposed that a definite diagnosis of PMA or PLS is only made >4 years after symptom onset to ensure that there has been no emergence of ALS signs.6 Even if patients exhibit solely UMN or LMN signs, often at biopsy there is evidence of more widespread neural loss, which would support the hypothesis that this is all the same condition. Although some distinction of the phenotypes is helpful to define prognosis, often the terminology can be confusing for patients. Subjective interpretation of the criteria may impact patients’ entry into clinical trials. Others have proposed alternative classifications to improve prognostic accuracy and to allow clearer description of the patient’s status for other clinicians and in clinical trials. Not only would this classification involve a description of UMN or LMN predominance and El Escorial category, but also diagnostic modifiers such as genetic mutation, presence of frontotemporal dysfunction, and stage of disease.6
Amyotrophic Lateral Sclerosis and Frontotemporal Dementia are on a Spectrum of TDP-43 Deposition Neurodegenerative Disease
There are conditions that have been associated with MND, such as FTD. Frontotemporal dysfunction occurs in up to 50% of ALS patients, reaching FTD criteria in 15%.9 TDP-43 has been found in both ALS and FTD. It is a RNA-binding protein which has been shown to be mislocalised in ALS patients. Restoration of the stathmin 2 gene (STMN2) allows microtubule stabilisation, and in turn allows recovery of axons in TDP-43-depleted motor neurons. This can indicate STMN2 as a target for future therapy.10 Some patients may also develop cerebellar or autonomic problems, possibly related to TDP-43 deposition in those areas of the nervous system. These associated features are not included in current clinical classifications. There is again controversy as to whether ALS–FTD is a separate entity compared to ALS with some executive dysfunction, because there appears to be differences in TDP-43 deposition and microglial activation.11 Furthermore, TDP-43 deposition is variable, and many ALS patients do not demonstrate obvious cognitive impairment, most notably Prof Stephen Hawking.
It is more easily conceivable to understand the TDP-43 deposition in the brain leading to UMN dysfunction. To what extent TDP-43 causes LMN dysfunction, such as in PMA, is unknown. Several pathological processes have been proposed in ALS, affecting both UMN and LMN. It may be that MND pathologies are numerous, and different emphasis on pathological mechanisms leads to the different symptomatology experienced.
Further clarification about the description of the diagnosis and phenotype has been proposed.6 This will assist in both research recruitment endeavours and being able to communicate information about diagnosis and prognosis
to patients.
Amyotrophic Lateral Sclerosis is Solely A Motor Neuronopathy
The concept of ALS only being a motor neuron condition has also been questioned. Not only have there been significant cerebral cortex changes mentioned above, there is recent research suggesting that sensory nerves are also affected.12,13 Somatosensory evoked potentials are also abnormal, suggesting sensory pathways in the spinal cord or brain are affected.14 Small-fibre neuropathy has also been demonstrated in ALS patients, though it is unclear how this correlates with phenotype and prognosis.15,16
Amyotrophic Lateral Sclerosis Can Be Split into Sporadic and Familial Subtypes
Approximately 10% of ALS cases have been described as ‘familial’.17 However, there remains significant numbers of so-called ‘sporadic’ cases which have something of a genetic basis.18 Contributory genetic variations and mutations are being identified, and their contribution to the pathway leading to ALS is gradually being understood.19 While cases such as those associated with SOD1 mutations follow an autosomal-dominant pattern, other heritable genetic traits may be involved in a less obvious manner in sporadic cases without obvious family history. The development of ALS has been shown to be a multi-step process involving both genetic and environmental risk factors.20 Family history itself may be difficult, since ALS is a disease of older age, and family members may have died before symptoms manifested. A positive family history is also affected by family size.21 Furthermore, penetrance of some genetic forms, such as the C9orf72 hexanucleotide repeat expansion, remains below 100%. The C9orf72 expansion is the most common genetic abnormality, found in approximately 40% of familial cases. It is also found in 4–8% of sporadic ALS cases (in a European and USA cohort).22 Approximately one-third of ALS and FTD will have a pathogenic C9orf72 expansion.23 Other relevant conditions such as FTD may not be diagnosed or recalled correctly when a family history is being sought. It has been shown that the risk of ALS in relatives of patients with sporadic disease is higher than in those without affected relatives.
Some now advocate a broader genetic screening for ALS patients with apparent sporadic disease.23 With the advent of antisense oligonucleotide therapy trials, identification of a mutation may lead to therapy. Greater adoption of genetic testing may also broaden the database of disease-causing mutations or expansions and increase our understanding of phenotypic presentations of different mutations. In the familial ALS cohort, different phenotypes and prognoses are already known: C9orf72 expansions are associated with more cognitive (FTD-type) deficits, whereas SOD1 mutations less so. Certain mutations may also confer a favourable prognosis.24
DIAGNOSIS
Current Diagnostic Criteria are Outdated and Do Not Include Consideration of New Diagnostic Techniques
Correct diagnosis of ALS is essential to counsel patients regarding their future. Other motor neuromuscular disorders, such as multifocal motor neuropathy (MMN), have effective treatments and are important not to miss. Early on in the disease course, however, it is often difficult to make a definite diagnosis of ALS; delays in diagnosis are common.25 The sensitivity of the revised El Escorial criteria may only be 57% at the time of diagnosis.26 Money and time can be spent on unnecessary treatments if the diagnosis is not made appropriately. First and foremost, diagnosis is made on clinical grounds, with supporting electrophysiological evidence. Imaging is used for exclusion of mimics, such a monomelic amyotrophy or multi-level polyradiculopathy.
The El Escorial Criteria (1994) were formed to standardise the diagnosis of ALS based on principally clinical parameters. It emphasised the need for UMN and LMN degeneration in the same region. There must be disease progression in the affected region, and there must be spread of degeneration to other body areas. It divides diagnosis into four categories: definite, probable, possible, and suspected. These criteria were revised in 1998.27 The principal change was to place more emphasis on electrophysiological evidence, allowing electromyography (EMG) evidence of denervation rather than clinical evidence, i.e., muscle wasting. Further recommendations were made in Awaji, Japan, in 2008 to facilitate earlier diagnosis.28
It allowed fasciculations in a chronically reinnervated muscle as evidence of LMN dysfunction along with fibrillations and sharp waves. This change increases diagnostic sensitivity by 12–20% on revised El Escorial criteria, without changing specificity.29-31
Diagnostic classification is useful for research purposes, but can be confusing when discussing the diagnosis with patients. For example, a patient may be defined by Awaji criteria as ‘possible ALS’, whereas the clinician is certain of the diagnosis based on the clinical presentation and exclusion of mimics. In a retrospective study of 399 standardised cases, there were large inter-rater variations in probable and possible categories.32 Some patients were classified differently between revised El Escorial and Awaji Criteria in the probable-laboratory supported and possible categories. There is an inherent complexity in interpretation of both clinical and electrophysiological parameters for the different body regions.
The World Federation of Neurology (WFN) subgroup on ALS produced an updated consensus statement in 2015 attempting to clarify the diagnostic classification.33,34 It took provided guidance on phenotypes not conforming to typical revised El Escorial criteria such as the flail arm or flail leg variant. Note was also made that there is a negligible false-positive rate in the possible cohort, and these can be considered for clinical trials (assuming appropriate exclusion of mimics). It also indicated that a pathogenic mutation in a known gene can substitute for UMN or LMN signs. These are all pragmatic recommendations recognising the heterogeneity of ALS and should allow easier access to clinical trials for patients earlier in the disease course. New consensus diagnostic criteria have recently been published which have sought to simplify the diagnosis of ALS by collapsing the previous definite, probable, and possible categories into a single diagnostic entity to facilitate clinical management and trial design.35
Diagnostic Modalities
Nerve conduction studies are necessary during ALS diagnostic workup for exclusion of mimics such as MMN (Table 1).36-38 Some further diagnostic information may also be gained from nerve conduction studies: increased H reflexes (relative to M wave) can indicate UMN dysfunction.36 A split-hand index can calculate the ratio of median compound muscle action potential (CMAP) compared to ulnar CMAP. A value <0.6 is indicative of the split hand phenomenon of ALS, and can be seen even when symptoms arise in another anatomical area.1 Additionally, a split-leg index can also be used.39 Unfortunately, CMAP amplitudes themselves are a blunt diagnostic indicator because they are preserved well into the disease course due to compensatory reinnervation. Motor unit number estimation provides more direct information about motor neuron loss, and several techniques have been developed for this. Some have the ability to distinguish ALS patients from controls at diagnosis.40 Yet given the variability at diagnosis, this technique may be more useful to monitor patients longitudinally, to monitor progress (more accurately than Revised Amyotrophic Lateral Sclerosis Functional Rating Scale [ALSFRS-R]) and provide prognostic information. Although one study demonstrated the MScanFit program to be most accurate for monitoring motor unit decline, this requires software that may not be available to all neurologists.41
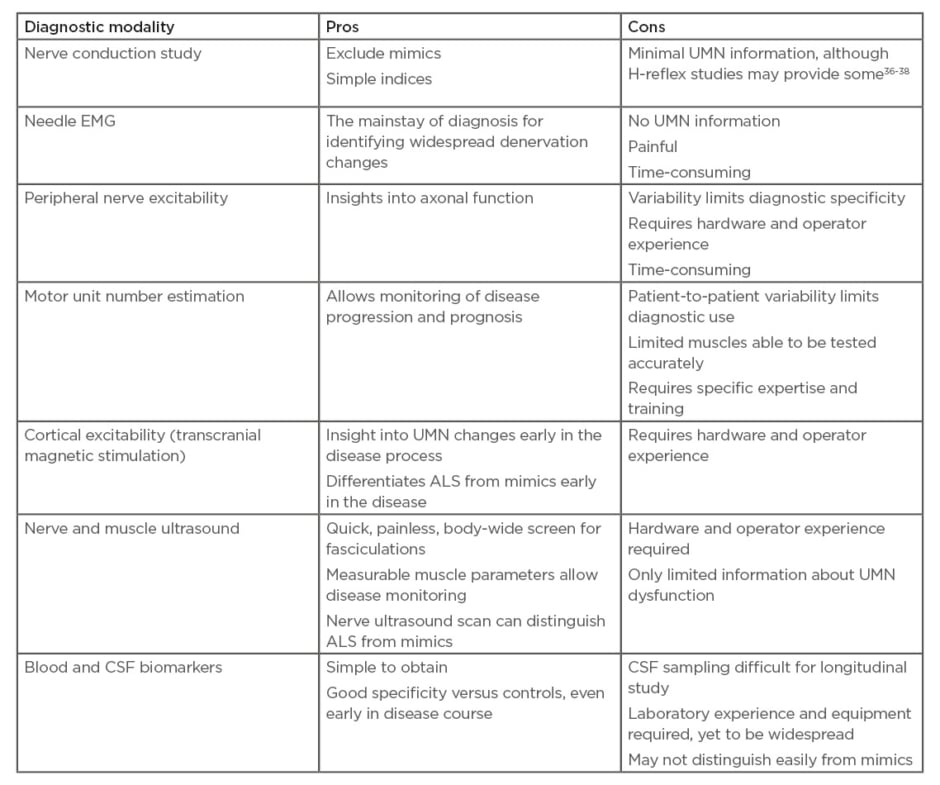
Table 1: Advantages and disadvantages of various diagnostic modalities.
ALS: amyotrophic lateral sclerosis; CSF: cerebrospinal fluid; EMG: electromyography; UMN: upper motor neuron.
It is Likely that Cortical Excitability Will Prove To Be a Useful Diagnostic Tool
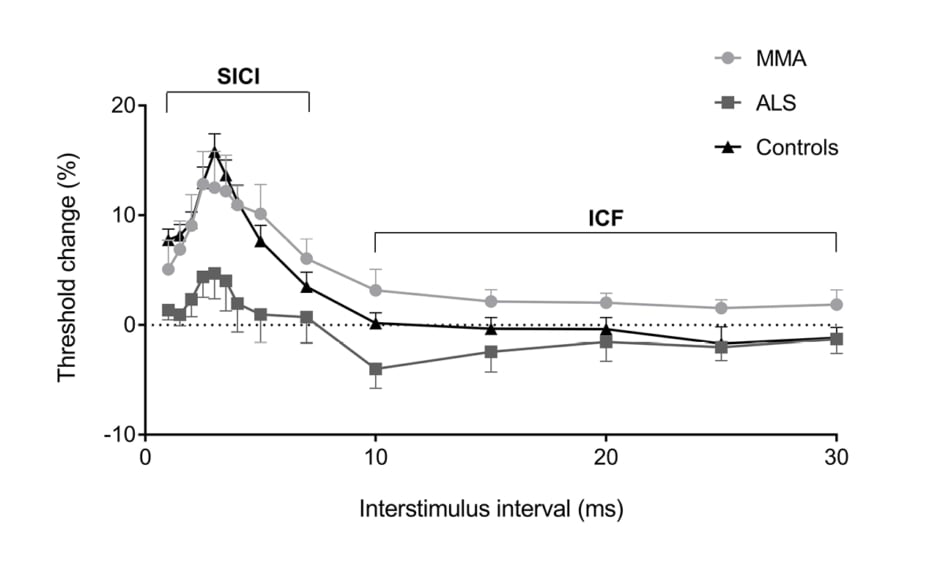
Motor evoked potentials (MEP) have been shown to be altered in ALS. MEP may be absent, decreased in amplitude, and may be delayed or dispersed (prolonged central motor conduction time [CMCT]).42 Absent MEP are seen in patients with more severe UMN dysfunction. Abnormal MEP may be helpful in differentiating ALS from a pure LMN disorder such as MMN; however, early ALS patients may have CMCT that fall within normal limits (Figure 1).43 Additionally, differentiating ALS from hereditary spastic paraparesis cannot be done on CMCT values, since both may be prolonged.44 The use of dual-stimulation transcranial magnetic stimulation (TMS) allows two magnetic impulses to be created in quick succession. How the cortex then responds to these impulses under different conditions, and whether a motor signal is generated, can provide insights into what is occurring in the cortex on a neuronal level.
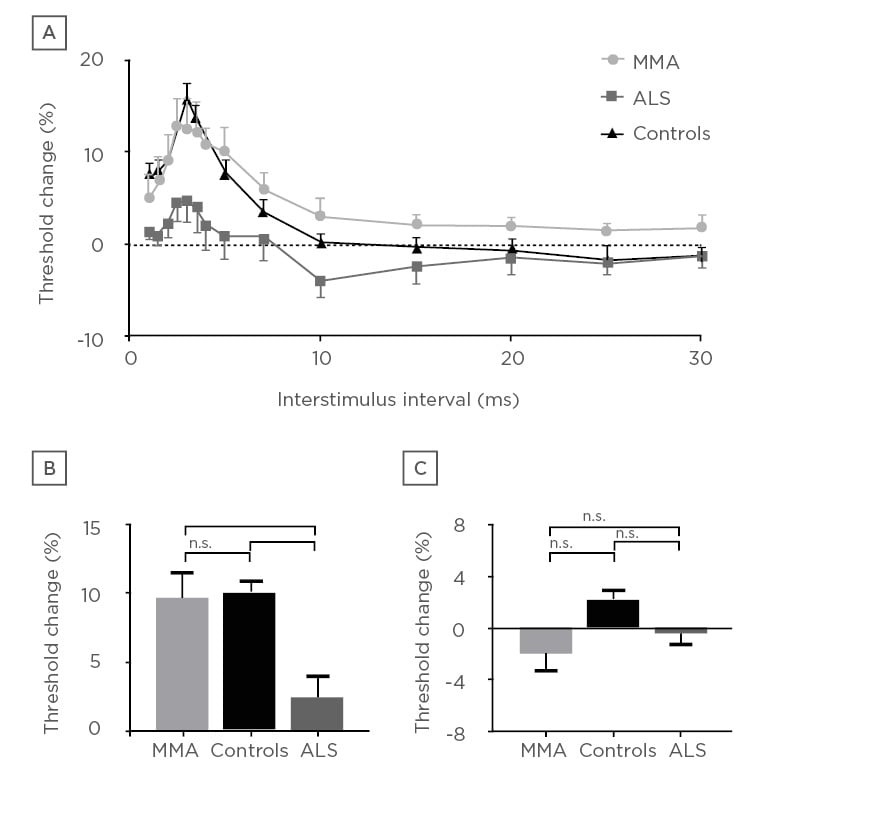
Figure 1: Cortical excitability in patients with monomelic amyotrophy compares with healthy controls and amyotrophic lateral sclerosis patients. Whereas the mimic (MMA) demonstrates normal cortical excitability, patients with amyotrophic lateral sclerosis demonstrate cortical hyperexcitability.
ALS: amyotrophic lateral sclerosis; MMA: monomelic amyotrophy; n.s.: nonsignificant.
*p≤0.005
***p≤0.001
Reproduced with permission from Matamala et al.43
Recent studies have focussed on cortical hyperexcitability as an integral component of ALS pathology. It is thought that cortical hyperexcitability occurs early in the disease process, and may result in anterior horn cell degeneration in a ‘dying forward’ process.45,46 It has been established that a normal motor cortex has a refractory period (absolute then relative) whereby a second motor signal cannot be created. This is known as the short interval cortical inhibition period (SICI). In patients with ALS, this SICI has been shown to be decreased. In other words, the cortex is more ready to generate a second motor impulse, only shortly after one has been sent, and is indicative of a hyperexcitable motor cortex. This reduced SICI is the most robust TMS diagnostic variable.47-49 This reduction in SICI occurs early in the disease process, occurring prior to symptoms in familial ALS50 and certainly when symptoms are subtle in sporadic cases.51 One series demonstrated that the number needed to test with TMS to diagnose an extra case of ALS was 1.8, demonstrating excellent diagnostic utility for the extra testing time. This was not affected by site of symptom onset.46 The degree of reduced SICI has been shown to be a prognostic marker for shorter life expectancy.52 A reduction in resting motor threshold and decreased cortical silent period duration have also been shown, consistent with cortical hyperexcitability for the symptomatic side of the body.47,48,53 These techniques can differentiate ALS from mimics,43 and forms part of a proposed ALS diagnostic index to improve early diagnosis.44,54 While the equipment may be expensive and requires operator experience, this will be a useful tool for earlier diagnosis.
Neuromuscular Ultrasound Complements Needle Electromyography and Should be Included in Future Diagnostic Criteria
Though not widely used when the El Escorial or Awaji criteria were devised, neuromuscular ultrasound is emerging as a useful diagnostic and prognostic test modality (Table 1).55 Its attraction comes from its noninvasive nature. It is also generally time-effective, being able to sample a wide variety of muscles in a short time.
For diagnosis, ultrasound can identify fasciculations easily, and can identify fasciculations in deeper muscles compared to needle EMG.56,57 It can also be used for initial screening of likely pathological muscles to increase needle EMG yield. The distribution of fasciculations may allow distinction of ALS compared to a benign fasciculation mimic or peripheral hyperexcitability syndrome.58 It has also been shown to facilitate differentiation of large fasciculations from myoclonus.59,60 However, it is inferior to EMG for defining the complexity of fasciculations, and fibrillations are better identified by needle EMG.61 Nevertheless, quantitative fasciculation analysis allowed differentiation from mimics with high sensitivity and specificity.57 Muscle ultrasound is particularly useful for bulbar evaluation, for which needle EMG of tongue muscles is painful and may be hindered by incomplete relaxation.61,62 The use of muscle ultrasound scores may help simplify use in diagnosis.63
Not only do muscles atrophy, their characteristics are changed as the disease progresses, with increased deposition of fibrous-fatty tissue. These changes can be quantified with ultrasound measures of echo intensity, echovariation, grey-level co-occurrence matrix, and increased muscle stiffness on elastography (Figure 2). These have been demonstrated to be useful adjuncts to clinical parameters in trials, and in some cases has shown to improve diagnostic accuracy compared to clinical tests alone.64,65
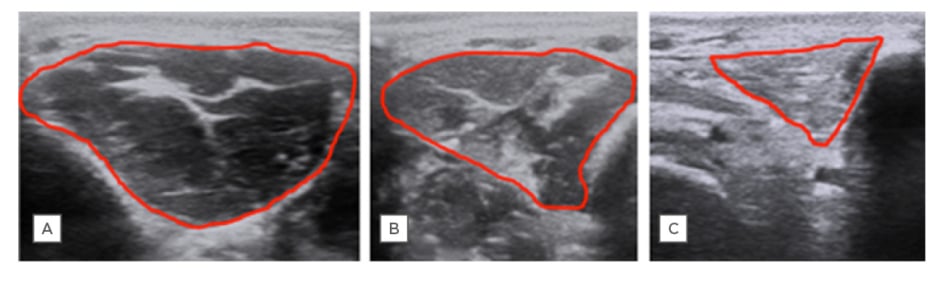
Figure 2: Ultrasound of the first dorsal interosseous muscle.
A) A healthy control; B) an ALS patient with minimal clinical hand weakness; and C) an ALS patient with severe hand weakness. Ultrasound demonstrates reduced muscle thickness/cross-sectional area and increased echogenicity with progressive denervation.
ALS: amyotrophic lateral sclerosis.
Ultrasound has also been used to identify bulbar muscle dysfunction related to UMN dysfunction.66 The ratio of muscle thickness from contracted to relaxed was shown to differ between bulbar ALS patients and controls. Further work is needed to determine how accurate this finding is on an individual level, and whether this can be added as a criterion for UMN deficit in the bulbar region. In a similar manner, thickness ratio changes of the diaphragm between inspiration and expiration have been used to identify weakness. This correlates with forced vital capacity, ALSFRS-R, and diaphragm CMAP. Diaphragm ultrasound is an attractive alternative to other measures because of its noninvasive nature and lack of requirement for good bulbar control to perform respiratory function tests.67 It should be noted, however, that diaphragm thickness changes are only one component of respiratory dysfunction in ALS patients, with intercostal and bulbar dysfunction as well as central drive impairment being important to consider as well.
As a disease progression monitoring tool, ultrasound has shown some promise, but demonstration of additional benefit to measures of disability, such as the ALSFR-S score, is required before it becomes useful, and this is still lacking.4
Nerve ultrasound has also been studied in ALS. Nerve cross-sectional area is reduced because of LMN loss. This differentiation can be used in clinical scenarios such as differentiating PLS (solely UMN) from UMN-predominant ALS, which shows nerve atrophy.68 Distal:proximal cross-sectional area ratios of peripheral nerves have been shown to reflect motor neuron atrophy typical of ALS and can be a diagnostic indicator.69 Other mimics such as MMN or chronic inflammatory demyelinating polyneuropathy often have enlarged nerves.
There is a building body of evidence to support the use of ultrasound in both diagnosis and monitoring of ALS patients. Some uses, such as fasciculation identification, are simply diagnostic. Other measures may not be useful in the early stages, such as muscle atrophy, but may be useful to monitor for disease progression and assist prognosis.
Standardisation of testing techniques for quantitative measures will be useful for both research and longitudinal patient monitoring. With machines improving in portability and cost, use of ultrasound is likely to become more widespread, and incorporation of ultrasound parameters in future diagnostic and prognostic paradigms is recommended.
MRI is a Useful Research Tool, But Adds Little to an Individual Diagnosis of Amyotrophic Lateral Sclerosis
MRI can be used for exclusion of mimics and is performed on most patients undergoing ALS diagnostic workup. Identification of corticospinal tract hyperintensity on T2-weighted imaging, as well as hypodensity on susceptibility-weighted imaging (SWI) in the motor cortex (the ‘motor band sign’) can be indicators for a diagnosis of ALS. One study identified the motor band sign in 78% of ALS patients on whom the SWI sequence was used, however numbers were small, and further research is required.70 With SWI or gradient echo sequences becoming standard, this may be a helpful piece in the diagnostic puzzle. Both of these MRI findings are nonspecific however, so should not be overemphasised.
MRI has been a useful research tool to evaluate the cortical changes seen in ALS. Voxel-based morphometry and diffusion tensor imaging have shown accelerated loss of grey matter, particularly in the premotor cortex. Changes appear to be most prominent in the ALS-FTD cohort.71 The corticospinal pathways are also disrupted. As the disease progresses, further loss is seen in frontotemporal areas outside the motor pathways.72 These techniques allow ALS patients to be differentiated from controls with a 65% sensitivity and 67% specificity for ALS. However, this is only on a group level and no definite conclusions can be drawn on an individual level.73
Blood Biomarkers Will Support a Diagnosis of Amyotrophic Lateral Sclerosis in the Future, and Can Be Used for Monitoring of Disease Progression
Neurofilament light chain (NfL) protein is emerging as a potentially useful biomarker in many neurological conditions. It is released from neurons undergoing axonal damage. Studies have shown increased levels in ALS patients compared to controls, but more importantly has shown the ability to differentiate from mimics.74-76 The titre has also been shown to vary with clinical stage, and therefore may aid in identifying disease progression. It appears to be higher in patients with UMN features and the level in cerebrospinal fluid (CSF) correlates with time to death, indicating use as a prognostic marker.74 Further work is required on this, however, because levels will vary on a patient-to-patient basis, and may also vary based on the site of disease manifestation. There is still an overlap between values in the ALS and mimic populations, therefore other diagnostic techniques should be used. Laboratory techniques have now improved to allow detection of NfL in the blood as well as CSF, and this will be important for its use to monitor disease progression, since serial CSF sampling is impractical. Though not as sensitive or specific as CSF for ALS diagnosis, serum or plasma levels still demonstrated good positive and negative-predictive values.75,76
Phosphorylated neurofilament heavy chain (pNfH) has also been studied and is similar to NfL with respects to sensitivity and specificity. A cut-off of 560 pg/mL yielded a sensitivity of 83% and a specificity of 80%. NfH did not change with disease progression in one small study, whereas a drop of NfH levels correlated with disease progression in another.77 A small study has also indicated that pNfH concentrations in the CSF can help differentiate UMN-predominant ALS from common mimics such as hereditary spastic paraparesis and PLS on a group level.78 There was significant overlap in groups on an individual level and therefore, again, this biomarker can only provide some supportive evidence for diagnosis. A limitation remains in the availability of a suitably validated immunofluorescence assay. Currently used at research centres, it may take time to become more widely available.
Numerous other blood and CSF biomarkers have been proposed for ALS.79 Given the relationship to FTD, CSF levels of TDP-43 have been tested in ALS patients. The CSF level could differentiate ALS patients from neurological controls with a sensitivity of 59.3% and specificity of 96.0%. The indication that lower levels of TDP-43 heralded a poorer prognosis, and that there was no correlation with disease duration, needs further investigation in a larger trial. Plasma TDP-43 levels have also been found to be higher in ALS patients than controls, although there was significant overlap of the two groups.80 Another study has shown that CSF metabolomics (eight selected metabolites) coupled with clinical parameters provided prognostic information in ALS patients.81 Using a proteomic method to identify CSF biomarkers, Thompson et al.82 identified three macrophage-derived chitinases that were more abundant in ALS patients and had some correlation with disease progression. Other CSF biomarkers of inflammation have also shown correlation with disease progression and some ability to delineate ALS cases from neurological controls.83 Whether these research techniques are able to be replicated in other labs, and can be used on a more general basis, remains to be seen. Furthermore, as with most tests, on an individual level there may not be sufficient separation between an ALS patient and a mimic.
TREATMENT
The role of the multidisciplinary team, and the use of symptomatic management including palliative care, are recommended for all ALS patients and are incontrovertibly beneficial. American guidelines were published on the topics of symptomatic treatment, nutritional support, and respiratory support in 2009,84,85 and similar guidelines exist in the UK (National Institute for Health and Care Excellence [NICE]) and Europe.86 Appropriate feeding and breathing adjuncts such as percutaneous gastrostomy and noninvasive ventilation should also be discussed with patient and family. These have been shown to prolong survival, and the only controversy arises as to when these adjuncts should be instigated. Commonly, this is a decision led by the patient and their family. Significant variation occurs with regards to the uptake of respiratory support, and this variation is sometimes unexplained though may relate to cultural and patient factors.87 The role of pharmaceutical agents for disease modification is more debatable.
Riluzole Should be Ceased in Later Stages of the Disease
Riluzole is a glutamate release inhibitor which has been used for more than 20 years. It has demonstrated tracheostomy-free survival benefit of between 2 and 3 months.88,89 It remains generally well tolerated, but may cause increased fatigue and transaminase abnormalities. Since the original studies, the extent of drug effect has been questioned, with only some study outcome measures being positive, and lack of cost-effectiveness limiting previous use in some jurisdictions.90,91 Some studies only identified a survival benefit in bulbar-onset rather than limb-onset ALS.92 Some showed bulbar-onset ALS to be more responsive,89,93 but this was contradicted in other studies.94 Its use at later stages of the disease is unclear and debated.94 Riluzole was shown to have an effect on cortical and peripheral axon excitability, but it was transient.95 Furthermore, studies suggest that riluzole is not effective following treatment for longer than 12 months.89,93
More recent retrospective trials, however, have indicated that riluzole works in advanced stages of disease, whereas the middle stages (King’s Stages 2 and 3) did not demonstrate a benefit.96,97 Given the possibility that riluzole is working in different ways at different stages of disease, some advocate continuing riluzole into the advanced stages and remains a preferred approach of many neurologists if the medication is tolerated.98,99
No Other Medications for Amyotrophic Lateral Sclerosis Have Been Shown To Be of Benefit
The antioxidant drug edaravone has been approved in Japan and the USA. Although the initial trial was negative,100 a subsequent randomised trial in a subgroup of rapid progressors was positive.101 Its use is limited by the requirement for intravenous infusion, its cost, and uncertainty regarding which patients will benefit. Other medications trialled have not demonstrated benefit, but hopefully there are more in the pipeline.102
Stem-Cell Therapy Represents the Future of Amyotrophic Lateral Sclerosis Treatment
As patients strive to find any effective therapy to slow or reverse their decline, they become susceptible to strong marketing for therapies which may be promising but have no evidence for benefit. Autologous stem-cell therapy (‘transplants’) has been used for immune reconstitution in multiple sclerosis and systemic sclerosis with good results, albeit with significant potential side effects. Patients are drawn to the idea of stem cells for its intuitive mechanism of action, and as a ‘nonpharmacological’ way of inducing their own body to repair itself. Private clinics are providing treatments to ALS patients with varying methods and standards.17 A recently published study with appropriately standardised protocols using autologous bone-marrow-derived mesenchymal stem cell transplant found a small benefit in a cohort of ALS patients demonstrating rapid disease progression.103 This was a Phase II study, and further studies are required before this onerous therapy becomes more mainstream. Patients should be encouraged to enrol in approved clinical trials to undertake experimental therapies, otherwise currently it is considered that potential harms of stem-cell therapy outweigh benefits.
CONCLUSION
This is an exciting time for ALS research, with understanding of pathological mechanisms increasing and diagnostic armamentarium evolving. Clinical trials for new therapeutics will come soon, and it is therefore important to improve diagnostic certainty early in the disease course for timely treatment. In this review, many of the uncertainties in the understanding of this heterogeneous condition have been demonstrated. It is clear that no single diagnostic test will identify all ALS cases. The diagnostic modalities described represent pieces of a puzzle, with each patient being sufficiently different to require a personalised diagnostic approach. The authors endorse the use of a proposed personalised diagnostic ALS index54 and propose the utilisation of these diagnostic techniques to be included in future diagnostic criteria. Specifically, they recommend the additional incorporation of cortical excitability and ultrasound evidence into laboratory-supported diagnosis in future consensus guidelines. Blood and CSF biomarkers may play a role if uptake is widespread, but greater certainty regarding diagnostic accuracy is required. Incorporation of genetic information (where available) should also be considered. From a therapeutics point of view, disease-modifying treatments are still some way off, and there remains controversy as to how beneficial, and in whom, the current treatments work. There is a need to adequately test therapies in a well-defined patient population prior to approval to ensure cost-effectiveness.