Abstract
Pontocerebellar hypoplasia (PCH) represents a heterogeneous group of congenital neurodegenerative diseases. Patients are afflicted by severe motor and mental impairments and most patients die at a young age. The hallmark of PCH is hypoplasia of the cerebellum and the pons, often in combination with supratentorial involvement. PCH is caused by autosomal recessive mutations in genes, most of which play a role in RNA metabolism. Twelve types (PCH1-12) are described, mainly based on clinical features. Identification of the responsible genes showed that the clinical classification did not match with the genetic classification leading to definition of subtypes. The authors expect that the increasing use of next-generation sequencing will lead to the identification of even more new PCH genes, widening the genetic and phenotypical spectrum. This will expand the classification and make it more complex. Therefore, the authors suggest that a new adjusted classification should be formulated to save the functionality of the PCH group.
INTRODUCTION
Pontocerebellar hypoplasia (PCH) describes a heterogeneous group of rare neurodegenerative diseases. PCH is characterised by hypoplasia of the cerebellum and pons with variable atrophy of supratentorial structures. The first in-depth neuropathological report of a PCH case was published in 1928.1 PCH was first described as a distinct disease group in 1993 by Barth with the classification of PCH1 and PCH2.2 The PCH spectrum was broadened to six PCH types over the following years based on phenotypes. RARS2 was the first gene to be associated with PCH and was reported in 2007.3 In the following years, the gene defects for the other five subtypes were reported.4,5 It also became clear that mutations in different genes could give a similar clinical PCH phenotype. The number of PCH (sub)types increased rapidly due to the introduction of next-generation sequencing, with a (sub)type classification for each new PCH related gene. Currently, the PCH spectrum describes twelve clinical PCH types with eleven genetic subtypes (MIM PS607596) (Table 1).5-27 All forms show an autosomal recessive inheritance pattern.
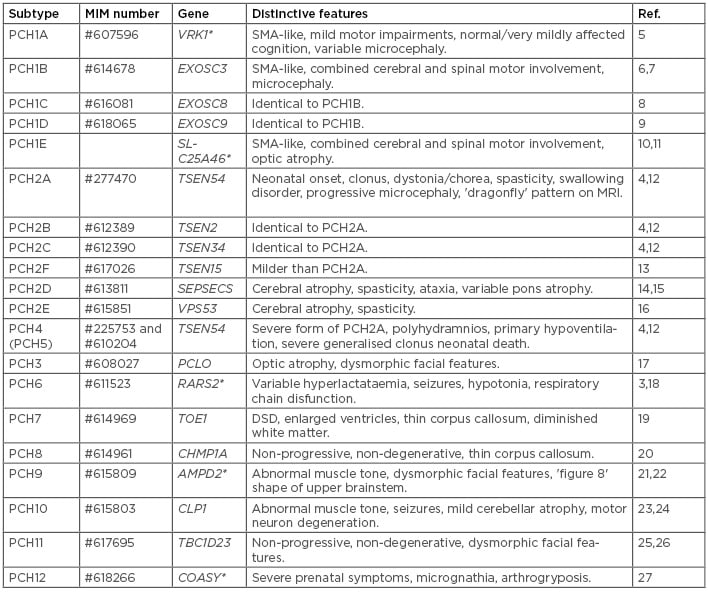
Table 1: Categorisation of the PCH group.
DSD: disorders of sexual development; PCH: pontocerebellar hypoplasia; SMA: spinal muscular atrophy.
*PCH (sub)types in which mutations in the involved gene also can result in another disease.
There is much clinical variability between different types but in general PCH presents as a severe disease with a prenatal onset or an onset of symptoms in the first months of life. Frequently, early symptoms are difficulties swallowing, apnoea, and respiratory difficulties indicating brainstem dysfunction. Prominent other symptoms are severe intellectual disability, epilepsy, and central motor impairment.4,12,28 Most PCH subtypes follow a progressive disease course and most patients die at a young age.12,29
Both hypoplasia and atrophy contribute to the reduction of cerebellar volume in PCH. Hypoplasia usually refers to a static state, while atrophy implies a progressive condition. However, it is often difficult to differentiate between the two mechanisms, as follow-up MRI scans are required to determine progression of cerebellar volume loss. PCH patients typically show severe hypoplasia of the cerebellar hemispheres with relative sparing of the vermis resulting in a typical ’dragonfly’ pattern on the coronal MRI, as seen in PCH2A patients (Figure 1e and Figure 1h). Another pattern is the ‘butterfly’; this reflects a milder form with less atrophy of the hemispheres (Figure 1b).12 Furthermore, patients display (progressive) supratentorial atrophy which underlies the microcephaly. In contrast to the gross infratentorial abnormalities, typical cerebellar symptoms are absent. This can be explained by the supratentorial involvement which hampers intentional motor skills. Below, this review will discuss the most common disease pathways in PCH, followed by a detailed description of the twelve types of PCH.
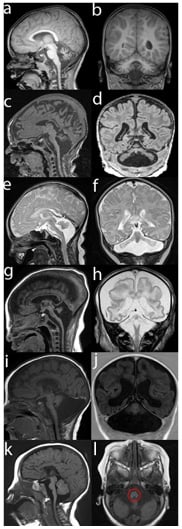
Figure 1: MRI images of PCH patients in sagittal (a, c, e, g, I, k), coronal (b, d, f, h, j), and axial (l) cross sections.
MRI ‘a’ and ‘b’ display a PCH1B patient with the mild phenotype correlating with the p.(Asp132Ala) change in the EXOSC3 gene. Other mutations in EXOSC3 are associated with more severe atrophy of pons and cerebellum (‘c’, ‘d’). PCH2A (‘e-f’) and PCH4 (‘f-h’), both caused by TSEN54 mutations, display the classical ‘dragonfly’ pattern seen on the coronal MRI (‘f’, ‘h’); however, in PCH4 more cerebral involvement is seen. TOE1 mutations cause PCH7 with severely dilated ventricles (‘i’, ‘j’). AMPD2 mutations cause PCH9 (‘k’, ‘l’), which show a typical ’figure 8’ pattern of the midbrain on the axial MRI (‘l’, red circle).
MECHANISM OF PONTOCEREBELLAR HYPOPLASIA
Although the pathology of different PCH subtypes is similar, there is no common pathomechanism for all of the PCH types; however, many genes are linked to different aspects of RNA metabolism (Table 1). This review will only focus on the most common pathway: RNA processing. EXOSC3, 8, and 9 (PCH1B-C) encode for components of the exosome complex; this complex is responsible for degrading different types of RNA, such as messenger RNA (mRNA) and transfer RNA (tRNA).6,8,9 TOE1 (PCH7) modifies specific RNA types called small nuclear RNA.19 The four components of the tRNA splicing endonuclease (TSEN) complex (PCH2A-C,F) together with CLP1 (PCH10) are involved in intron removal from tRNA.4,13,23 Mutations in these genes possibly trigger stress pathways caused by accumulation of aberrantRNA fragments in the cells. Activation of stress pathways can suppress protein synthesis leading to neurodegeneration.
Another possible mechanism is shortage of properly processed tRNA, which are responsible for connecting the correct amino acid to the corresponding trinucleotide code on the mRNA in the ribosomes during translation.23 There are 417 human tRNA genes while there are only 20 different amino acids. Despite the vast redundancy in tRNA genes, the lack of one specific tRNA gene can already cause progressive cerebellar atrophy in mice.30 Almost 7% of the tRNA genes contains an intron;31 thus, it is conceivable that tissue-specific expression of intron containing tRNA metabolism genes can result in dependence of the cerebellum on the tRNA splicing machinery. Other tRNA-related PCH genes, such as RARS2 (PCH6) and SEPSECS (PCH2D), are responsible for the production of tRNA coupled to their corresponding amino acids.3,14 However, it remains unclear why these aberrations in tRNA metabolism cause a brain specific defect.
The remaining PCH-related genes are thus far not functionally groupable and vary widely in mechanistic functioning.
PONTOCEREBELLAR HYPOPLASIA 1
PCH1 is hallmarked by anterior horn cell degeneration (as seen in spinal muscular atrophy) in addition to PCH. The severity of the disease course of PCH1 in patients is varied. PCH1 is often neonatal lethal because of respiratory insufficiency and severe hypotonia. Congenital contractures and polyhydramnios can also be present at the severe end of the PCH1 spectrum. In milder cases however, the pons is spared, and patients survive into adolescence. So far, five subtypes have been described (PCH1A-E).6 PCH1B (MIM #614678), caused by EXOSC3 mutations, is the most prevalent subtype of PCH1.32 A PCH1B patients’ disease course can be mild or severe, depending on the genotype.7 A homozygous p.(Asp132Ala) change in EXOSC3 is associated with sparing of the pons and a mild disease course (Figure 1a–b): patients display some cognitive development and limited speech. Other EXOSC3 mutations are associated with a more severe disease course, which is characterised by arthrogryposis and respiratory insufficiency which is often lethal before the first year of life (Figure 1c–d).6 EXOSC8 and its paralogue EXOSC9, also components of the exosome complex, underlie PCH1C (MIM #616081) and PCH1D (MIM #618065), respectively. The phenotypes of patients with EXOSC8 or EXOSC9 mutations are similar to those of EXOSC3 patients.8,9In 2016,
Wan et al.33 reported mutations in SLC25A46 in patients with PCH1E. Later, the authors of this present review showed that the PCH1 patients, described by Barth (1993), were compound heterozygous for mutations in SLC25A46.2,10 The clinical spectrum of patients with SLC25A46 mutations is very broad, ranging from death within the first postnatal month, to survival into adulthood and with patients becoming symptomatic later in life, presenting with mild cerebellar atrophy and axonal peripheral neuropathy.10,11,33 Notably, all patients have optic atrophy in this subtype.34 Mitochondrial disorganisation and enlargement was detected in cells of PCH1E patients, which is in line with the function of SLC25A46 in mitochondrial fission.10 Lastly, PCH1A (MIM #607596) is caused by mutations in the VRK1 gene and is sometimes called spinal muscular atrophy with PCH. It is a rare and atypical subtype often reported in patients with an Ashkenazi Jewish origin. Patients seem to present no mental impairments but do display a delayed motor development or only slow, progressive loss of motor functions later in life.35
PONTOCEREBELLAR HYPOPLASIA 2
Of the six PCH2 subtypes (PCH2A-F) that are currently described, PCH2A (MIM #277470) is the most common. Typically, neonates show jitteriness, clonus, poor sucking, and impaired swallowing. Later extrapyramidal movement disorders such as dystonia and chorea, spasticity, and the lack of intentional motor skills become apparent.36 Patients often are afflicted by generalised seizures, sleeping disorders, and chorea which often necessitates institutional care. Respiratory insufficiency and infections are a common cause of death which is generally at an early age, however some patients reach adulthood.29 The neuroradiological hallmark for PCH2A is the ’dragonfly’ pattern on MRI of the hypoplastic cerebellum (Figure 1e–f). In patients with a follow-up brain MRI, progressive cerebral atrophy is seen, with apparent ventricular enlargements accentuated by disproportionate affection of the caudate nuclear heads.12 Neuropathological findings in PCH2A showed stunted folial growth, especially affecting the cerebellar hemispheres, and peculiar fragmentation of the dentate nuclei.37 A breakthrough was achieved in 2008 with the discovery that mutations in the TSEN54 gene underlie PCH2A. The mutation, p.(Ala307Ser), was initially found in a Dutch genetic isolate and is the most common cause of PCH2.4 Patients with the same mutation p.(Ala307Ser) are also found on other continents suggesting that it is a very old mutation. TSEN54 forms the TSEN complex with three other components: TSEN34, TSEN2, and TSEN15. Mutations in TSEN2, TSEN34, and TSEN15 are very rare and result in PCH2B (MIM #612389), PCH2C (MIM #612390), and PCH2F (MIM #617026), respectively.4,12,13 One patient with a TSEN34 mutation and four patients with TSEN2 mutations have been reported.12,38 TSEN15 mutations are described in three PCH2F patients and have a milder phenotype with no visual impairments.13 The two other PCH2 subtypes are not caused by aberration in the TSEN complex, namely PCH2D (MIM #613811) and PCH2E (MIM #615851). Both were previously described in literature as progressive cerebrocerebellar atrophies. PCH2D is caused by mutations in the 0-phosphoserine tRNA-selenocysteine tRNA synthetase (SEPSECS) gene. Reported features of PCH2D are spasticity, optic atrophy, ataxia, and seizures. The MRI shows progressive cerebellocerebral atrophy with variable involvement of the pons.14 PCH2D disease course is usually severe; however, a mild PCH2D phenotype has been recently reported.15 PCH2E, caused by VPS53 mutations, has been reported in twelve Jewish families of Moroccan origin.16,39 All cases carried the same mutation, p.(Gln95Arg), suggesting a founder effect. Most patients appear healthy at birth but quickly develop hypotonia and delayed head growth. Subsequently, patients develop tonic-clonic -seizures, progressive spasticity, and progressive cerebrocerebellar atrophies on brain MRI.16
PONTOCEREBELLAR HYPOPLASIA 3
Only one family, from Oman, with PCH3 (MIM #608027) has been reported. All four affected children have a homozygous nonsense mutation in the PCLO gene.17 Generalised hypotonia with hyperreflexia, progressive microcephaly, and seizures developed during their first year of life. The patients reported thus far all survived into puberty. MRI shows an atypical pattern with diffuse atrophy of the cerebellum and pons; however, not as drastic as a ’dragonfly’ pattern. Supratentorial cerebral reduction of white matter with a thin corpus callosum was reported. PCLO is thought to play a role in regulation of proteins in neuronal synapses.17
PONTOCEREBELLAR HYPOPLASIA 4 AND 5
PCH4 (MIM #225753) describes a severe form of PCH2. Notably, patients present prenatal symptoms such as intrauterine seizures and polyhydramnios resultant of impaired fetal swallowing.40,41 Neonates show massive unprovoked myoclonus, hypertonia, hypoventilation, and sometimes seizures.12 Patients usually require continuous mechanical ventilation and generally die within the first month of life. Brain MRI shows subtotal destruction of cerebellar hemispheres, which are underdeveloped and atrophied with often mild-to-severe vermal folia atrophy (Figure 1g–h). In addition, cerebral atrophy is more apparent than in PCH2 (Figure 1g–h). Craniocerebral disproportion with a vastly increased subarachnoid space may be seen as the result of progressive loss of cerebral tissue with a prenatal onset ([hlFigure 1h[/hl]). Post-mortem examination revealed only remnants of the dentate nuclei and the inferior olive nucleus had a horseshoe shape, therefore PCH4 was originally named ‘olivo-ponto-cerebellar-hypoplasia’.37 Similar to that seen in PCH2A, TSEN54 mutations also underlie PCH4 (MIM #225753). PCH4 patients are often compound heterozygous for the common p.(Ala307Ser) mutation in combination with a loss of function mutation. Although patients with homozygous null-alleles of TSEN54 have not been described, the authors assume that this is incompatible with life. PCH5 (MIM #610204) was initially described as a distinct subtype based on the presence of intrauterine seizure-like activity in one family.41 In retrospect, PCH2A, PCH4, and PCH5 represent mutations of the same gene where increasingly severe symptoms commensurate with the severity of mutation.42
PONTOCEREBELLAR HYPOPLASIA 6
Mutations in the autosomal gene encoding the mitochondrial argenyl tRNA synthetase (RARS2) cause PCH6 (MIM #611523). Characteristics of PCH6 include severe early onset seizures, CSF hyperlactataemia, mitochondrial respiratory chain defects, and later severe developmental delay with rapidly progressive microcephaly.3 Neuroradiologic imaging reveals a global progressive atrophy of the brain with variable hypoplasia/atrophy of the cerebellum.18,43 The basal ganglia were spared in some patients with severe cerebral atrophy. Neuropathological reports of two cases reveal an intact olive nucleus and relatively spared dentate nucleus.43
PONTOCEREBELLAR HYPOPLASIA 7
PCH7 (MIM #614969) is characterised by disorders of sexual development in addition to PCH.19,44 Clinical symptoms are truncal hypotonia with hypertonic extremities, seizures, and increased tendon reflexes. Most described patients present with female external genitalia. Ten XY-patients are reported with a small or absent penis and undescended or absent testis. An ultrasound revealed ovaries in two of these patients. In one of the three XX-female patients, no ovaries could be found using ultrasound. Female external genitalia were normal; although one patient showed a prominent clitoris. A brain MRI revealed severely reduced cerebellar and pontine volumes with enlarged ventricles, diminished white matter, and a thin corpus callosum in some patients (Figure 1i–j). Patients carried homozygous or compound heterozygous mutations in the autosomal TOE1 gene. The function of TOE1 remains elusive, although current evidence points to a role in snRNA metabolism in the nucleolus.19
PONTOCEREBELLAR HYPOPLASIA 8
CHMP1A mutations are rare and underlie PCH8 (MIM #614961).20 Only three families and two different pathogenic mutations have been reported. In contrast to the other types of PCH, the disease course of PCH8 appears to be non-progressive. Patients have microcephaly, dystonia, and/or choreiform movements, and sometimes suffer from joint contractures and seizures. Developmental delay is severe, but some patients learn to walk and develop limited speech. Brain MRI shows PCH with reduced cerebral white matter volume and a thin corpus callosum. In patients with follow-up brain MRI, no progression of the atrophy was visible. Mechanistically, it is thought that CHMP1A aberrations disrupt the BMI1regulated proliferation of neuronal progenitor cells.20
PONTOCEREBELLAR HYPOPLASIA 9
PCH9 (MIM #615809) describes a broad disease spectrum with 25 affected individuals reported to date. Clinically, patients have progressive microcephaly, impaired swallowing, brisk tendon reflexes, spasticity, teeth abnormalities, and some patients display dysmorphic facial features.21,22,45 While some patients survive into adulthood, most die early in life. In addition to the cerebellar hypoplasia, PCH9 patients displays a distinctive ‘figure 8’ shape of the midbrain on axial MRI (Figure 1l). Also, a hypoplastic or absent corpus callosum has been reported. Mutations in the AMDP2 gene are found in PCH9, but are also identified in one family with hereditary spastic paraplegia 63 (SPG63)46 (MIM #615686). Aberrations in AMDP2, which regulates GTP levels, is thought to disrupt the initiation of protein translation.
PONTOCEREBELLAR HYPOPLASIA 10
PCH10 (MIM #615803) patients show dysmorphic facial features, cognitive impairment, seizures, and spasticity.23,24 Brain MRI showed only mild atrophy of the cerebellum and the pons. Supratentorially, atrophy of the cortex, and a thin hypoplastic corpus callosum are reported. Furthermore, signs of progressive spinal motor loss are seen on electromyography. All PCH10 patients originated from Turkey and carried the same homozygous mutation in the CLP1 gene, suggesting a founder effect. The CLP1 protein interacts with the TSEN complex and is involved in tRNA processing.23,24
PONTOCEREBELLAR HYPOPLASIA 11
PCH11 (MIM #617695) is reported in seven families who all carry biallelic mutations in the TBC1D23 gene. Individuals are relatively mildly affected compared to other PCH types, and so far, a non-progressive nature of PCH11 is noted.25,26 Patients present with dysmorphic facial features, microcephaly, hypotonia, and occasional ambulation. Brain MRI shows a reduced cerebellar volume and severe hypoplasia of the pons. The corpus callosum is hypoplastic in most cases. TBC1D23 is involved in neuronal vesicular trafficking.25
PONTOCEREBELLAR HYPOPLASIA 12
The latest branch of the PCH tree is PCH12 (MIM #618266). PCH12 is a lethal disorder at the severe end of the PCH-spectrum, presenting with severe prenatal onset microcephaly, PCH, and congenital contractures.27 Atrophy of the cerebrum and hypoplasia of the pons, cerebellum, brainstem, spinal cord, and basal ganglia were confirmed on postnatal brain MRI in one of the patients. Genetic analyses identified splice site and frameshift mutations in the COASY gene in affected individuals. COASY is responsible for the last two steps of coenzyme A (CoA) synthesis; the CoA synthesis pathway is highly conserved and essential for life. Earlier reports have also linked COASY mutations to neurodegeneration with brain iron accumulation 6 (MIM #615643) (NBIA6). Residual function of COASY appeared to be higher in NBIA6 patients than in the one tested PCH12 patient, which might explain the more severe phenotype in the latter.47
DIFFERENTIAL DIAGNOSIS
Several other diseases should be considered when a patient presents with PCH. The three most common diseases that mimic PCH phenotypes are caused by CASK mutations, PMM2 mutations, and extreme prematurity. Mutations or microdeletions in the CASK gene cause mental retardation and microcephaly with pontine and cerebellar hypoplasia (MIM #300749) highly similar to PCH. In addition, neocortical malformations, hearing loss, and facial dysmorphic features can be present.48 Overall, development delay is severe, but some patients reach some motor milestones, such as walking. In contrast to all PCH types, CASK mutations follow a X-linked dominant inheritance pattern and mutation are generally de novo.49 Moreover, patients with heredity disorders of glycosylation type 1a (PMM2-CDG) (MIM #212065) can mimic PCH. Neuroimaging of PMM2-CDG patients reveals cerebellar hypoplasia/atrophy.50 Clinically, the phenotype of PMM-CDG patients varies from mild intellectual disability and ataxia to a severe phenotype with antenatal onset multi-organ abnormalities and severe mental and motor impairments. Distinctive dysmorphic features include inverted nipples and abnormal fat distribution. PMM2-CDG can be diagnosed by detection of abnormal glycoproteins through isoelectric focussing of transferrin. Additionally, extreme prematurity (gestation age <32 weeks) can occasionally result in PCH.51 The rapid growth of the cerebellum during the third trimester might increase vulnerability to disruption by very early prematurity.
When lissencephaly is noticed on brain MRI in addition to PCH, mutations in RELN (MIM #257320), VLDLR (MIM #224050), or α-dystroglycan related dystrophies should be considered.52,53 Furthermore, patients at the severe end of the Hoyeraal-Hreidarsson syndrome (MIM #30500) spectrum and spinocerebellar ataxia 29 (SCA29) (MIM #17360) can resemble PCH. Patients with Hoyeraal–Hreidarsson syndrome usually present with a combination of progressive bone marrow failure and cerebellar hypoplasia.54 Furthermore, a patient with an ITPR1 mutation, normally resulting in SCA29, showed classical PCH on brain MRI.55
DIAGNOSIS OF PONTOCEREBELLAR HYPOPLASIA
The diagnoses of PCH is based on careful clinical examination and neuroimaging, followed by genetic testing. Genetic testing is required to confirm the diagnosis because differentiation between subtypes of PCH and other disorders is often not straightforward. Furthermore, for most PCH types the number of patients described is low (e.g., PCH3 or PCH8), and sometimes limited to a single mutation (e.g., PCH10), leading to an incomplete phenotypical characterisation.15,56 Therefore, the notion should be taken that PCH phenotypes can deviate from reported descriptions.
PERSPECTIVES
PCH is a clinical diagnosis in patients with atrophy and hypoplasia of the pons and cerebellum. Unlike the suggestion implied by the name PCH, they represent a group of disorders that affect development of the structure and function of the whole brain. Progressive decline in structure and progressive microcephaly characterise the most common types. Identification of many different PCH genes and types resulted in a rapidly expanding classification. Initially described as a disease associated with defects in (t)RNA metabolism, the identification of CHMP1A and VRK1 mutations challenged this hypothesis.5,20 The classification became more complex when cases with milder disease manifestations, such as PCH1B and PCH10, were included.7,23 This resulted in the current PCH classification as a collection of disorders that do not always share a common phenotype or affected pathway (Figure 2). Even the criteria of cerebellar and pontine atrophy are not always met; therefore, the authors propose that the classification should be revised, reserving the name PCH for the clinical description of patients with early onset atrophy of the cerebellum and often pons. The most obvious classification is based solely on clinical features, i.e., PCH with disorders of sexual development (PCH7), PCH with dystonia (PCH2), and PCH with spinal muscular atrophy (PCH1). Moreover, the disease course should be considered in the classification because it is important for counselling. The lack of knowledge about pathomechanisms in most forms currently precludes a classification based on mechanism. The broad phenotypic spectrum within patients with mutations in the same PCH gene also precludes a purely genetic classification; therefore, the clinical symptoms and MRI should be leading in the diagnosis and the genetic diagnosis can be used for counselling and disease course prediction.
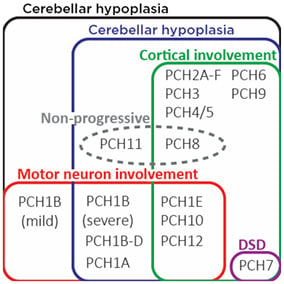
Figure 2: Depiction of pathological overlap between different PCH types.
In PCH2A-C, F, patient cortical involvement is not always present at birth but will always develop postnatally.
DSD: disorders of sexual development; PCH: pontocerebellar hypoplasia.
CONCLUSION
PCH remains a severe disease for which only symptomatic treatment is available. Preconception carrier screening in high-risk populations is successful and feasible for some subtypes.57 It is likely that in the future more genes will be linked to PCH and new mutations in known PCH genes will broaden the phenotype further.