Abstract
Non-dystrophic myotonias (NDM) are rare muscle disorders caused by mutations in skeletal voltage-gated muscle channels leading to delayed muscle relaxation after voluntary contraction. They are subdivided into sodium channelopathies, when the mutation is in the SCN4A gene, and chloride channelopathies, when the mutation is in the CLCN1 gene. Symptoms, which may differ according to subtype, exacerbating factors, and over disease course, can include muscle stiffness, pain, fatigue, muscle hypertrophy, myalgia, and weakness. The severity of NDM symptoms varies widely, from being barely noticeable to causing considerable disability that impacts health-related quality of life. People with NDM may remain undiagnosed for several years, potentially due to a lack of awareness of NDM among many healthcare professionals. The symptomatic treatment for NDM predominantly involves sodium channel blockers, such as mexiletine. Randomised, placebo-controlled trials have shown mexiletine can reduce muscle stiffness and pain, and improve health-related quality of life. Patient and clinician surveys, as well as national guidelines, place this medication as one of the first choices for pharmaceutical treatment of myotonia. Other choices include lamotrigine, carbamazepine, acetazolamide, ranolazine, and flecainide, though clinical evidence is limited, and all are used on an off-label basis. Herein, the challenges in recognising and treating myotonia symptoms in people with NDM are reviewed, along with strategies to increase awareness of the disease and its potential treatment.
INTRODUCTION
Non-dystrophic myotonias (NDM) are a group of rare muscle disorders with a reported prevalence of 0.75−1.7 per 100,000 population (Figure 1).1,2 They are due to pathogenic variants in genes coding for skeletal voltage-gated muscle channels that cause ion channel dysfunction. These mutations lead to increased muscle membrane excitability,3 clinically manifesting as delayed muscle relaxation after voluntary contraction or percussion.4-6 NDM symptoms and signs can include muscle stiffness, cramps, myalgia, transient weakness, fatigue, muscle hypertrophy, dysphagia, and dysphonia.4 Depending on NDM subtype and exacerbating factors, these may differ in age of onset, body distribution, severity, and circumstances of occurrence.4,7-9 Symptoms can lead to movement difficulties, such as in running, climbing stairs, or grip release after a handshake.6,9 While NDM shares similarities with myotonic dystrophies, muscle weakness is typically more common in myotonic dystrophy Type I than NDM. There are overlapping phenotypes with myotonic dystrophy Type 2, but cold-exacerbation of symptoms is highly suggestive of paramyotonia congenita (PMC) and people with myotonic dystrophy Type 2 may have temperature-sensitive pain and myotonia.8
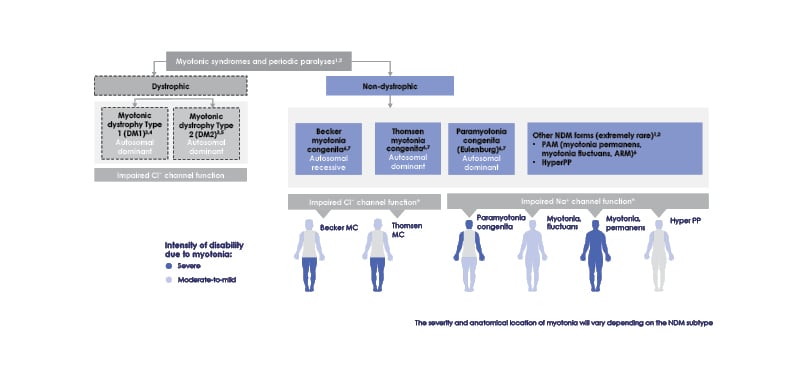
Figure 1: Non-dystrophic myotonia subtypes.
*Due to wide variability in affected regions experienced with all types of non-dystrophic myotonia, location of affected muscles is not an accurate indicator of NDM subtype.
ARM: acetazolamide responsive myotonia; Cl-: chloride ion; HyperPP: hyperkalaemic periodic paralysis;
MC: myotonia congenita; Na+: sodium ion; NDM: non-dystrophic myotonia; PAM: potassium-aggravated mytonia.
Chloride channelopathies (NDM-ClC) arise due to mutations in the chloride channel CLCN-1 gene, and include the autosomal dominant Thomsen myotonia congenita (TMC)10 and the autosomal recessive Becker myotonia congenita (BMC).11 Typically starting in childhood (TMC) or adolescence (BMC), both are present with myotonia that decreases or vanishes with repetitive motion. BMC tends to be more severe than TMC and is associated with episodic and occasionally progressive weakness.4,6,11,12 Triggers in both conditions can include cold, psychological stress, exercise, and alcohol consumption.7
Sodium channelopathies (NDM-NaC) are due to autosomal dominant SCN4A gene mutations affecting the NaV1.4 channel, and include sodium channel myotonia and PMC, as well as hyperkalaemic periodic paralysis and severe neonatal episodic laryngospasm. With a typical onset around age 5 years, symptoms differ according to diagnosis but can include episodic weakness, pain, and isolated facial stiffness. These can be exacerbated by cold temperatures, potassium consumption (depending on subtype), menstrual cycle, and, for PMC especially, repetitive contractions. While weakness may be present, it is most prevalent in hyperkalaemic periodic paralysis and can last for days.4,12,13
Although there are many shared symptoms in NDM, patient surveys reveal that clinical and percussion myotonia, stiffness, muscle hypertrophy, and pain may be more prominent in NDM-ClC, with muscle weakness, hand grip myotonia, paradoxical eyelid myotonia, cramping, and cold exacerbation more prevalent in NDM-NaC.7,8,12,14 However, this does not always hold true, with one patient survey finding subjective muscle weakness reported by 75.0% of people with NDM-ClC and 36.7% of those with NDM-NaC.15
To understand experiences of NDM diagnosis, symptoms, treatment, health-related quality of life (HRQoL), and support, two surveys were carried out among patients with NDM and their caregivers. The 2020 IMPACT (Impact of non-dystrophic Myotonia on PAtients and Caregivers’ qualiTy of life) patient (n=181) and caregiver (n=59) surveys included responses from several European countries and the USA.16,17 The 2018 European Myotonia Observation Survey of Patient Access to therapy avoiding Harm (MyoPath) survey involved 390 people with a variety of myotonic disorders and included 43 people with NDM (data on file, Lupin Neurosciences).18 Results of a roundtable expert panel discussion on the IMPACT survey findings are also included herein. This involved seven Europe-based clinicians experienced in managing adults with NDM.
DIAGNOSIS OF NON- DYSTROPHIC MYOTONIA
While NDM typically first manifests in childhood, symptoms may be mild and not recognised as burdensome, resulting in diagnostic delay and misdiagnosis.9 For instance, a difference in gait or fall frequency may be dismissed as normal for the spectrum of childhood movement.19 Patients themselves may not recognise NDM symptoms that have been present since infancy or are not perceived to impact their life.3 The expert panel also highlighted how families affected by similar symptoms might not notice them as being unusual. Conversely, as NDM can be familial, symptoms may be more readily recognised by family members.5 Symptoms may, however, be aggravating, debilitating, and progressive or, in TMC, first appear or worsen during pregnancy.9,20
In the IMPACT survey, diagnostic delay was reported by 64% of respondents, who experienced symptoms for ≥10 years before diagnosis.16,17 Similarly, a German study found average delays of 15−17 years.7 The IMPACT survey showed that diagnostic delay might be due to incorrect referral to a specialist not experienced enough in NDM to recognise the symptoms, including a general neurologist, orthopaedist, or rheumatologist, instead of to a neuromuscular specialist.17 The expert panel also discussed how patients may experience difficulties in describing their symptoms, and a discrepancy between the language used by patients and healthcare professionals (HCP) might lead to misunderstanding and incorrect referrals.
Education to raise awareness of NDM among HCPs to help ensure patients receive a correct and timely diagnosis was a need highlighted by the expert panel. They emphasised the necessity for organisational structures that included a high level of collaboration among stakeholders to refer and diagnose patients quickly. The panel also suggested a need for a red flags list for easier NDM recognition, such as myotonia, myalgia, and, in some cases, increased serum creatine kinase levels.
NDM diagnosis involves observations of NDM signs and symptoms, as well as neurological and neurophysiological examinations and genetic testing.4 Action myotonia can be detected by asking the patient to open their eyes after holding them tightly closed for a few seconds or by assessing grip release ability following a firm handshake. Percussion myotonia can be elicited in the muscles using a reflex hammer.4-6 The presence of exacerbating factors, which can vary both between patients and over time, can be assessed via a symptom diary.21 Family history is also useful in those with autosomal dominant NDM subtypes such as TMC.4-6
NDM can also be assessed using physical ability measures with best test–retest agreement being shown for the Timed Up and Go test,22 Borg’s Category-Ratio Scale,23 and the Myotonic Behaviour Scale,24 along with patient-rated symptom scales and 14-day stiffness diaries.25 However, few of these tests have been specifically validated to measure myotonia severity or other NDM symptoms, which contributes to diagnostic delay and suboptimal assessment of disease symptoms.
HRQoL can be measured using the Individualised Neuromuscular Quality of Life (INQoL) measure, a patient-completed questionnaire that includes four domains regarding how symptoms affect HRQoL, how NDM limits life domains, how treatment affects symptoms, and overall HRQoL.26
Testing of muscle function in NDM involves needle electromyography (EMG) to ascertain myotonic discharge presence in affected muscle fibres (i.e., electrical myotonia), which may be present without clinical symptoms. EMG has a high sensitivity for myotonia detection; however, detection of electrical myotonia is not specific for NDM and, in the case of a positive test, other disorders, such as myotonic dystrophies, need to be eliminated. The short exercise test, at room temperature or after cooling, helps distinguish between NDM-NaC and NDM-ClC.5,14,25 The combination of EMG with compound muscle action potentials changes after short or long exercise tests produces distinct Fournier patterns that might help toward differentiating NDM subtype.14,27 EMG testing cannot, however, assess symptom severity,5,25 and may not be generally available.
Although costly, the only way to definitively confirm NDM subtype is with gene panel testing.4 Muscle MRI can reveal NDM-related muscle abnormalities, such as fatty changes and oedema, that are distinct from those shown in other myotonic disorders, but is not a useful tool for diagnosis.27,28 Future research with muscle MRI is needed to ascertain its use in distinguishing patterns of involvement or monitoring treatment response.
NEGATIVE EFFECTS OF NON-DYSTROPHIC MYOTONIAS ON HEALTH-RELATED QUALITY OF LIFE
Overall, people with NDM report an inferior health status and HRQoL compared with control populations.15,29 This may be related to specific symptoms such as muscle stiffness, pain, and fatigue.12,15,29 For example, in a patient study,15 over a quarter of patients with NDM-ClC and over half with NDM-NaC characterised myotonia as painful, and reported that it negatively affected general health perception, physical and social functioning, and vitality. Around half of respondents experienced fatigue, which inversely correlated with vitality and physical health scores.15
A study utilising the INQoL questionnaire (n=66) found particular impacts of weakness, ‘muscle locking’, pain, and fatigue. Such impacts were evidenced in daily life, work, and leisure activities as well as on independence. Scores were slightly higher (greater impact) for those with NDM-NaC. This study also showed that 6.3% of patients with NDM-ClC and 23.5% of those with NDM-NaC were disabled due to their condition, and 3.1% and 14.7%, respectively, were unemployed due to NDM symptoms.12 Another study utilising this measure also found greater HRQoL impacts for people with NDM-NaC with greatest impacts on activity limitations, independence, and emotions.14
Another important finding of the IMPACT survey was that over a quarter of respondents rated their HRQoL as low. Participants reported that NDM interfered with social, practical, physical, and work or study related realms, in areas such as communicating with others, using public transport, undressing, and carrying out their chosen profession. Emotional impacts of NDM were also reported, with many respondents saying that they felt helpless, isolated, and alone.16,17 Further, 42% of respondents in the IMPACT caregiver survey reported negative impacts on their mental health and 25% on their physical health.17
MANAGEMENT OF NON-DYSTROPHIC MYOTONIA SYMPTOMS
Pharmacological treatment for NDM primarily centres around sodium channel blockers. In the IMPACT survey, of those who had received treatment, symptoms reduced by such medications included muscle stiffness, pain, and tiredness.17 However, 41% of respondents had not received NDM-specific medications.16,17 The reasons for this are multifactorial and include the perception that symptoms are mild, poor awareness by patients and HCPs regarding treatment options, unavailability of some medications in some countries, and reluctance to start treatments that are also indicated in non-NDM conditions.3
The expert panel agreed that they might not offer medication to patients whose symptoms are described or perceived as minimal or appear non-significant and where HRQoL is not perceived to be impacted. They may also not offer medication to patients with comorbidities, especially cardiac problems, a history of drug allergies, previous treatment failure, or pain explained by other factors. They discussed how the decision to initiate medication might be patient-led, based on how much they can tolerate their symptoms versus their concerns regarding potential treatment-related adverse events (AE). It was noted that patients are usually more likely to want treatment following specialist clinic consultation.
Table 1 details several of the current treatments for NDM and provides some guidance for switching from off-label medications to mexiletine, which has among the highest level of evidence using in NDM. Mexiletine, a Class 1B antiarrhythmic sodium channel blocker, is the only European Medicine Agency (EMA) approved antimyotonic drug for NDM. It reduces sodium influx, allowing faster inactivation and repolarisation of sodium channels.4 As mexiletine is less potent than other antiarrhythmic sodium channel blockers, and recovery from sodium channel binding is faster, it has a safer cardiac action compared to Class IA and Class IC sodium channel blockers.37
NDM does not affect the cardic muscle,38 as cardiac muscles do not significantly express the NaV1.4 sodium channel isoform affected in NDM,39 and very few CLC-1 chloride ion channels.40 The effectiveness of mexiletine in managing symptoms, including muscle stiffness, pain, and weakness, and improving HRQoL, has been shown in several trials (Table 2).21,41-43 Due to its actions on the heart,44 as with any antiarrhythmic drugs for managing myotonia, cardiac evaluation prior to prescription and annually/every 2 years is a requirement for administration.45 For mexiletine, while in one randomised controlled trial one patient had asymptomatic bradycardia that resolved without study withdrawal on follow-up ECG,41 in other studies, no ECG conduction abnormalities were reported.21,42,43 Mexiletine administration has not been shown to lead to QT interval prolongation in people with a healthy heart.37
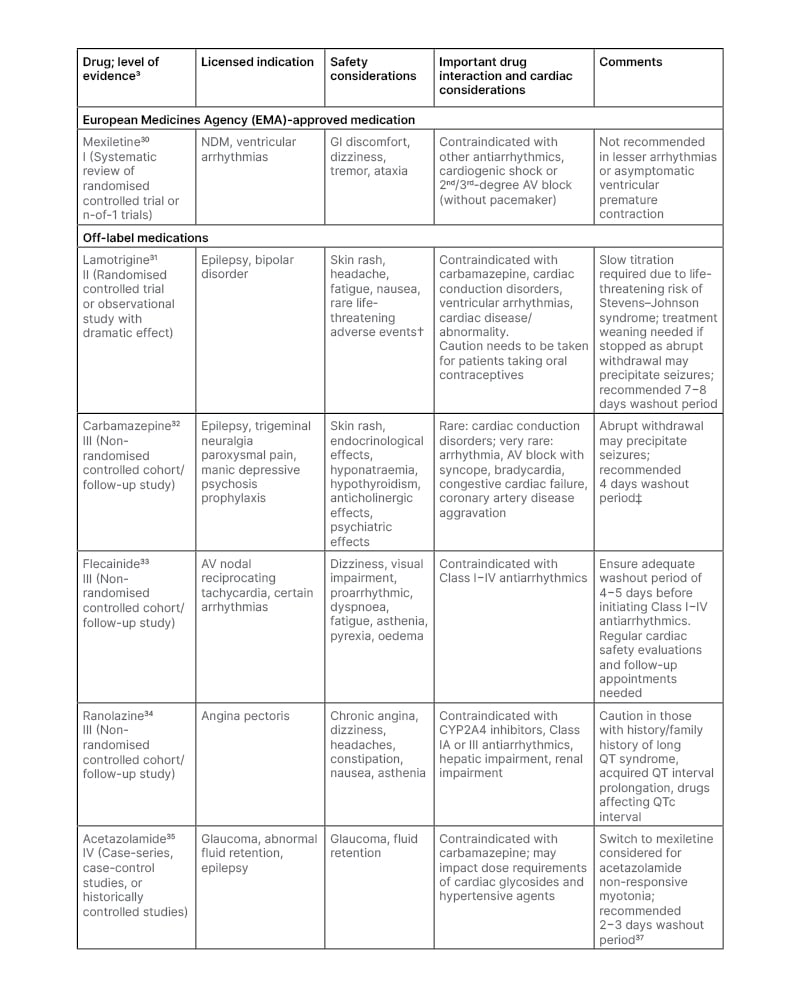
Table 1: Medications utilised for the treatment of myotonia in non-dystrophic myotonias.
†Stevens–Johnson syndrome, toxic epidermal necrolysis, aplastic anaemia, bone marrow depression, pancytopenia.
‡Based on 5.5 elimination half-lives to clear a medicine from the system.
AV: atrioventricular; CYP: cytochrome P450; GI: gastrointestinal; NDM: non-dystrophic myotonia.
The most common AEs in mexiletine studies were gastrointestinal.21,41-44 In a long-term study of 63 patients (mean follow-up: 4.8 years [range: 0.5−17.8 years]), this predominantly manifested as dyspepsia, leading to treatment discontinuation in four patients.43 No clinical studies have been carried out regarding mexiletine use during pregnancy, though case studies have described the successful use of such.46
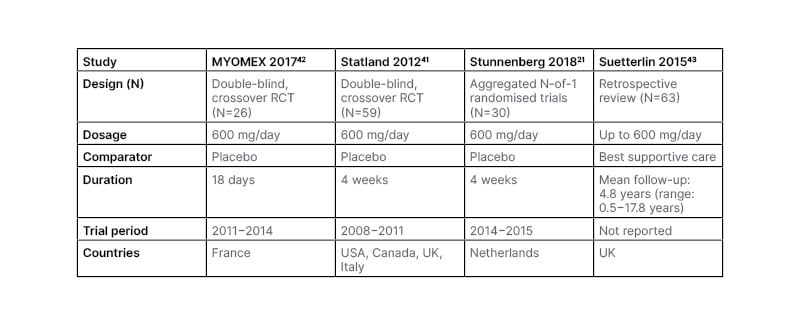
Table 2: Mexiletine studies.
RCT: randomised controlled trial.
Most of the expert panel considered mexiletine as first-line treatment for NDM in patients without contraindications. However, challenges to prescribing mexiletine included unavailability of cardiologists to conduct the cardiac assessment, uncertainties of the cardiac problems associated with mexiletine, and access issues such as reimbursement cost and paperwork burden. In another study, a panel of experts was convened to investigate healthcare utilisation regarding mexiletine. They discussed how treatment with mexiletine led to decreased need and frequency of use of NDM-related resources including physiotherapy, occupational therapy, mental health support, and walking stick use.47
The MyoPath survey was conducted to maintain the orphan drug designation of mexiletine by the EMA.48 The outcomes discussed an improvement in muscle stiffness and increased mobility in patients with NDM receiving mexiletine. Patients also reported a positive impact on HRQoL domains including emotional wellbeing, confidence in their abilities, and simple daily tasks. Treatment interruptions worsened myotonia and were associated with fatigue, pain, dysphagia, breathing difficulties, impaired digestion, and poor sleep quality. Anxiety about mexiletine’s availability was expressed by 36/54 (67%) of currently-treated patients. Lack of mexiletine awareness was also reported, but this was an expected finding as MyoPath was undertaken before mexiletine was approved for NDM in 2018 (data on file, Lupin Neurosciences).18
Another treatment prescribed for NDM symptom alleviation is the sodium channel blocker lamotrigine, though its use is off-label (Table 1). Lamotrigine use for NDM is supported by a placebo-controlled, crossover trial (n=12 with PMC; n=14 with myotonia congenita) where administration of up to 300 mg/day for 8 weeks reduced MBS scores by 29%. Significant between-group differences favouring lamotrigine were also shown in a Timed Up and Go test, a 14-step stair test, hand and eye relaxation tests, and overall patient self-assessment of health (including physical and social functioning and role limitations due to physical health). AEs included headache, skin rash, fatigue, and muscle pain.31,49 The expert panel described how they would prescribe lamotrigine first line for symptomatic patients with cardiac-related mexiletine contraindications or an abnormal ECG; hepatic problems; co-existing depression, generalised/focal epilepsy, or mood disorders; intolerance to mexiletine; had altered potassium/magnesium blood levels; or were planning a pregnancy. The recent U.S. Food and Drug Administration (FDA) warning on the potential increased risk of arrhythmias with lamotrigine was noted by the panel.50
Several other medications are used for NDM treatment, though all are off-label (Table 1).4 Precautions need to be taken when switching between medications due to potential cardiac AEs. Flecainide, a Class IC antiarrhythmic drug, was viewed by the expert panel as more widely available and nearly as effective as mexiletine in those with a suitable NDM subtype. While it is considered the preferred first line treatment for severe neonatal episodic laryngospasm,51 efficacy has only been shown via case reports.52 Limitations though include the potential for occurrence of arrhythmias and an increased incidence of cardiac disease with prolonged use.3,53 The antianginal drug ranolazine can reduce EMG-detected myotonia and patient-reported stiffness as well as decrease weakness and pain. Mild light headedness and constipation were reported as AEs.54,55
Carbamazepine is a voltage-gated sodium channel blocker that was a choice expressed by the expert panel after failure of mexiletine, lamotrigine, or lacosamide, with only case reports available to assess use in NDM.3,56,57 Also used by the expert panel in some NDM subtypes was acetazolamide if other treatments were not well-tolerated or effective. An observational study of use in NDM (n=9) showed that administration decreased myotonia. AEs included paraesthesia and, in one case, potentially, renal calculus,58 though this has not been shown in other studies.59 According to the expert panel, lacosamide was used if mexiletine and lamotrigine failed; however, to date there have been no randomised controlled trials of its use in NDM.
In a survey including 70 patients with NDM, the three most prescribed drugs were mexiletine, carbamazepine, and flecainide, with fewer patients taking lamotrigine or acetazolamide along with several other antimyotonic drugs. The greatest patient perceived efficacy scores were given to mexiletine and lamotrigine.7 Another survey, including 37 people with NDM who took mexiletine, lamotrigine, ranolazine, or acetazolamide, revealed that lack of efficacy and AEs were the main reasons for medication discontinuation of any of these medications.8
The IMPACT survey revealed that 29% of respondents had not received any type of treatment for NDM and 34% were not satisfied with how their condition was managed.16,17 Both the IMPACT17 and MyoPath (data on file, Lupin Neurosciences)18 surveys highlighted the symptoms that patients most wanted improved included overall mobility problems, muscle stiffness, fall frequency, persistent tiredness, and pain. This was mirrored by a need for a reduction in HRQoL impacts.
Historically, for those with NDM symptoms defined as mild, practical help and advice to avoid triggers and exercise according to NDM subtype was the main therapeutic approach recommended by the expert panel; however, due to the nature of the condition, such avoidance is not always possible.3,12,60 They suggested that drug treatment for myotonia be offered to all patients with NDM even if they report symptoms to be mild, or the HCP perceives them as such as many will have had symptoms since childhood and may not be aware of how much they are interfering with their daily lives.3
The expert panel discussed several solutions for improving NDM management, including discussions with stakeholders, multidisciplinary approaches, multicentre collaboration, and further data collection. Multidisciplinary teams were seen as key to NDM management to help facilitate a holistic and individualised approach to care. The exact team composition may vary according to patient need but could include a neuromuscular specialist, physiotherapist, pain specialist, rheumatologist, and psychologist.
UNMET NEEDS
While understanding and treatment of NDMs are progressing, there are still unmet needs. For example, a study of patient-reported outcomes in NDM highlighted a need for a treatment strategy that targets mutation-specific biophysical abnormalities to better control symptoms. This would require greater understanding of how the affected ion channels function.12 The mechanism of pain within NDM is also poorly understood.60 As pain may be refractory to treatment, and is associated with HRQoL impact,15 it is important to elucidate the molecular pathways involved to tailor treatment approaches.60
Overall, the expert panel highlighted a need to increase awareness of NDM and confidence in recognising and treating this condition, as well as fostering a multidisciplinary approach to NDM care. Development needs to be undertaken of verified diagnostic tools and disease monitoring tools such as smartphone applications. The panel also suggested building on the findings of the IMPACT survey to further investigate what other medications patients with NDM were taking, including painkillers, to assess if treatment leads to a reduction in pain symptoms. Additionally, the panel wanted to understand how satisfied patients were with their current treatment and how caregivers were burdened and suggested that patient-reported outcome measures and HRQoL scales, potentially via smartphone applications, could be used to measure and monitor the effectiveness of drug treatment on symptoms over time.
With regard to increasing awareness of myotonia treatment, while most of the expert panel did not think mexiletine had a negative safety profile, they highlighted the need for pharmacodynamic studies to better understand mexiletine’s mechanism of action as well as patient registry studies to capture long-term efficacy and safety. This would help in the discussion with patients and other HCPs regarding the use of such therapy. They also discussed a need for further understanding of cardiologists’ opinions on long term use of mexiletine and other off-label treatments, and to clarify whether such medications are associated with cardiac risks in the NDM setting, as some panel members reported cardiac issues associated with mexiletine, lamotrigine and carbamazepine. A working group between cardiologists and NDM specialists has been formed in France, with the intention to publish the outcomes of the project.
The IMPACT survey revealed that fear of AEs was one reason given by patients who were not taking pharmaceutical treatments. This, the expert panel agreed pointed to a need to increase patient understanding of the advantages and disadvantages of antimyotonic medication, and align on treatment expectations.
Currently, mexiletine is only indicated for adults with NDM, as clinical studies did not include children. To address this, an 8-week clinical trial is underway including participants aged 6−17 years old. This will involve a 4-week titration phase up to age-appropriate bodyweight doses of mexiletine.61
CONCLUSION
Symptoms of NDM may differ according to NDM subtype and exacerbating factors, and may progress over time. While for some people symptoms may not impact daily aspects of life, for others, they can be disabling and severely disruptive.12,16,17 Although there are several pharmacological treatments utilised for NDM, only mexiletine effectiveness has been demonstrated in a number of clinical trials of NDM,21,41,42 and is the only approved drug for NDM,30 with lamotrigine effectiveness demonstrated in a small clinical trial.49 The expert panel highlighted how unmet needs include validated screening and monitoring tools, HCP education regarding NDM diagnosis and treatment, and multidisciplinary teams to tackle physical, emotional, and practical aspects of NDM.