Abstract
Duchenne muscular dystrophy (DMD) is a severe and fatal muscle condition affecting young children. Without interventions, affected boys lose the ability to walk independently by the age of 10 and develop progressive cardiac and respiratory failure. The last 20 years have seen a change in the natural history of DMD following improvements in clinical care and proactive interventions to manage complications of the disease. An international collaboration of DMD experts has created care imperatives for best practice in DMD; these are now available in 30 different languages and are disseminated worldwide. An update of these care recommendations is currently under review.
More recently, the field has seen encouraging scientific progress in regard to new therapeutic approaches of which a large number are currently being evaluated in clinical trials. With time, improvements in clinical care and access to new treatments and innovations are changing the natural course of DMD, from a relentless progressive illness with death in teenage years to a more chronic illness with a good quality of life and increased life expectancy. This is a particularly encouraging time for DMD, and experiences built in the muscular dystrophy field are likely to be of benefit to the development of new approaches and therapies in other rare diseases.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most severe and common muscular dystrophy in children affecting approximately 1 in 3,500–10,000 male births.1 It is an X-linked recessive condition affecting mostly boys. Clinically, it presents with progressive muscle weakness and wasting affecting all body muscles, including the heart and the respiratory muscles. Historically, boys with DMD lost the ability to walk independently by the age of 10 years and progressively developed heart and respiratory insufficiency, eventually leading to death in their late teens or early 20s. In recent years, there have been significant developments in both clinical care and scientific research in DMD. Following the implementation of care recommendations, including corticosteroids, respiratory and cardiac management, orthopaedic intervention, and rehabilitation, the natural history of DMD has changed from a rapidly progressive and life-limiting condition to a more chronic illness with an increase in life expectancy.2-3 Recent studies have reported a good quality of life,4 with the research field of DMD thriving, and the hope of finding a disease-modifying drug which will further slow the deterioration in muscle function. In this article, we summarise recent developments in the diagnosis and management of DMD and provide an update on clinical research in the field.
DUCHENNE MUSCULAR DYSTROPHY
Pathogenesis
DMD is caused by mutations in the dystrophin gene, the largest gene in the human genome, consisting of 79 exons. The mutation rate of the dystrophin gene is relatively high and about one-third of cases are caused by new (de novo) mutations.5 In the remaining two-thirds of patients, the mutation is passed on from the mother (X-linked), who usually does not show any clinical manifestation of the condition (asymptomatic carrier). The size of the gene, together with the high mutation rate, explains the variability of mutations in DMD including large deletions (68%), duplications (11%), and small mutations (20%).5 Small mutations include small deletions, insertions, and point mutations, resulting in stop codons or disrupted splicing.
The hotspot for mutations in the dystrophin gene lies between exon 45 and 55 (deletions) and exons 2 and 10 (duplications), but mutations have been reported along the entire gene. There are no strict correlations between the location of the mutation and the severity of the clinical presentation, although several exceptions to this general rule have now been described.5-8 Mutations causing an interruption of the ‘reading frame’ (out-of-frame) lead to DMD because protein synthesis is disrupted, resulting in the total absence of the encoded protein, dystrophin. Mutations in the same gene which do not alter the reading frame (in-frame mutations) lead to the production of a shorter but still partially functioning protein and result in a milder phenotype (Becker muscular dystrophy).
The protein dystrophin is expressed in skeletal muscles, the heart, and the brain. In muscles, dystrophin acts as a shock absorber during muscle contraction; in its absence, muscle fibres undergo membrane fragility, inflammation, chronic damage, and finally replacement by fibrotic tissue and fat. These pathological changes manifest clinically with progressive loss of muscle strength and mass. Although the role of dystrophin in the brain remains largely unknown, intellectual impairment is reported in about 30% of DMD subjects.9
CLINICAL FEATURES
The hallmark of DMD is progressive muscle wasting and weakness. Although the pathological changes start in utero, the condition does not usually manifest clinically until the age of 2–4 years. It can present with a delay in motor milestones or with symptoms of muscle weakness. Typical early clinical features of DMD include difficulty getting up from the floor (Gower’s manoeuvre), difficulties with running, jumping, hopping, an abnormal or tip-toe gait, and frequent falls. On examination, calf hypertrophy, hypotonia, and neck flexor weakness can be detected from an early age.10 Speech and language delay and behavioural problems, including autistic spectrum disorders, have also been described and can present before muscle weakness has become apparent. With the progression of the disease, muscle weakness affects all skeletal muscles, leading to loss of independent walking, reduced motor function in the upper limbs, and scoliosis due to axial weakness. Joint contractures result from a combination of reduced mobility and changes in muscle structure, following replacement of muscle fibres with connective tissue. The heart and the respiratory muscles undergo similar pathological changes, leading to progressive dilated cardiomyopathy, arrhythmia, and restrictive lung disease.10
Early heart involvement has been reported in DMD, although generally the incidence of cardiomyopathy increases with age.11 The clinical challenge remains recognising heart failure in DMD, which often is asymptomatic, even at late stages. Respiratory insufficiency in DMD is more predictable, usually presenting after the loss of independent ambulation. The diaphragmatic involvement significantly contributes to the drop in forced vital capacity, observed as an early sign of respiratory impairment, increasing susceptibility to chest infections, and sleep-disordered breathing.12,13
DIAGNOSIS
If there is a clinical suspicion of DMD, a blood test to measure creatine kinase (CK) levels should be promptly arranged as the first screening test. CK is a muscle enzyme which is released in the blood stream as a response to muscle damage. CK levels are always significantly elevated in DMD, and values in the thousands should trigger the diagnosis, although other neuromuscular diseases might also be considered. CK testing has been used for neonatal screening, as increased levels are present from birth. CK newborn screening was in place in some countries e.g. Wales and the USA, however at present it has been discontinued due to concerns regarding its sensitivity and specificity when performed around the time of birth.14-16 At the time of writing, CK neonatal screening is not routinely adopted in any country.
A child with clinical evidences of muscle weakness and elevated CK levels should be referred to a neuromuscular centre for genetic testing for dystrophin gene mutations. Multiplex ligation-dependent probe amplification17 is the most reliable and cost-effective test to detect deletions and duplication, analysing all exons of the dystrophin gene. If multiplex ligation-dependent probe amplification does not identify any mutation, gene sequencing will allow the identification of small deletions/insertions and point mutations, although it is more expensive, time-consuming, and available only in specialised laboratories.
Muscle biopsy, which historically represented the diagnostic test for DMD showing the total absence of dystrophin expression in muscle fibres associated with utrophin upregulation, is today often avoided and replaced by genetic testing in centres where this is available. A muscle biopsy can still be useful in some rare cases when genetic testing does not reveal a mutation but the patient shows a clear DMD phenotype or when there is a discrepancy between the reading frame rule and the clinical presentation.
Delays in Diagnosis of Duchenne Muscular Dystrophy
Despite the tremendous advances in the management of DMD for children over the last decade, published data show that there is still a delay in the diagnosis of the condition across different countries.18-23 This is due to both presentational delay (time from onset of symptoms to presentation to a health professional) and diagnostic delay (time from first medical assessment to genetic diagnosis). The mean age at diagnosis ranges from 4.3–4.11 years and has not significantly improved over the past 20 years.18-23 Recommendations to lower the age of diagnosis include raising awareness of DMD in the community, primary care to ensure DMD is recognised as a differential diagnosis in children presenting with signs of muscle weakness and/or speech delay, and promoting early CK level testing as a cost-effective, readily available screening tool for DMD. To reach these targets, a mnemonic (MUSCLE: Motor milestone delay, Unusual gait, Speech delay, CK asap, Leads to Early diagnosis of DMD) has been developed for use in primary care to improve the diagnostic process in DMD.18
Establishing an accurate genetic diagnosis of DMD at a young age is important as it allows genetic counselling and family carrier testing, prenatal diagnosis, early access to treatment, consideration for mutation specific therapeutic options (e.g. ataluren), or participation in clinical trials with new investigational drugs.
MEDICAL MANAGEMENT
Corticosteroids
Corticosteroids are currently the only medication available to all patients with DMD to slow down the progression of the condition and are part of the care recommendations. Although the benefits of corticosteroids are well-documented, they only postpone disease milestones without altering the natural course of the disease. Moreover, corticosteroids are associated with several side effects which limit their prescription worldwide.
Corticosteroids have been shown to prolong ambulation and stabilise respiratory function.24-27 In addition, corticosteroids have cardio-protective properties and prevent or reduce the need for spinal (scoliosis) surgery.25,28,29 The most frequently prescribed corticosteroids in DMD are prednisone (or prednisolone) (0.75 mg/kg/day) and deflazacort (0.9 mg/kg/day), with several regimes currently used.30 Corticosteroids are generally introduced when the child’s motor skills plateau, around the age of 4–5 years. Emerging evidence suggests that early treatment might be associated with better long-term outcomes, including prolonged ambulation, however further research is needed to evaluate the impact of early and prolonged corticosteroid treatment in a growing child.31
Although corticosteroids have been prescribed for >20 years in DMD, a worldwide debate is ongoing as to which corticosteroid should be prescribed, which regime, from what age, and for how long. The main concerns are related to the management of steroid-related adverse effects which have led to inconsistency in prescribing practices across different centres and different countries. The more common steroid-related side effects and their management strategies according to the care recommendations are summarised in Table 1.3 Daily regimes appeared to be more effective but with more frequent side effects compared with intermittent regimes.32 Limited data comparing the effects of different corticosteroids are available: there are some indications that deflazacort is associated with less weight gain and possibly a better effect on muscle strength and function, but longer term longitudinal data are awaited.33-35
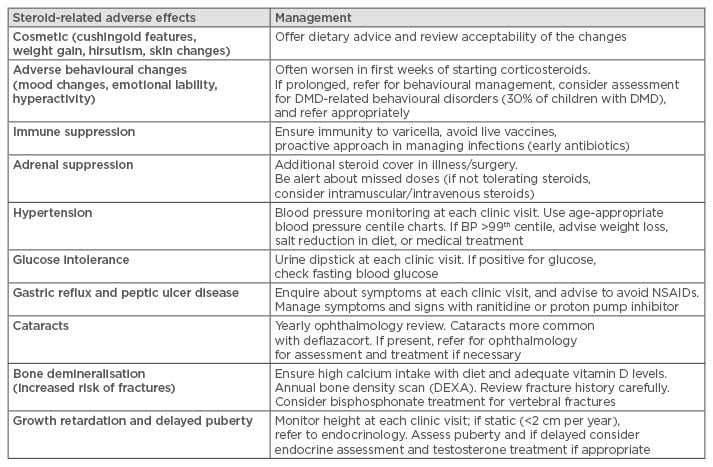
Table 1: Summary of adverse effects of corticosteroid therapy and their management.
DMD: Duchenne muscular dystrophy; BP: blood pressure; NSAIDs: non-steroidal anti-inflammatory drugs; DEXA: dual energy X-ray absorptiometry.
A double-blind randomised control study FOR DMD is ongoing to compare the three most commonly prescribed corticosteroid regimes in DMD regarding efficacy and side effect profile (NCT01603407).35 The study aims to clarify the long-standing equipoise of the use of corticosteroids in DMD, and provide families and clinicians a (long-awaited) evidence-based recommendation on the use of corticosteroids and standardised management of steroid-related side effects. Whilst awaiting the outcome of this study, the choice of treatment is currently made based upon personal experience of the clinician in charge, availability of the formulation (e.g. deflazacort is not available in the USA), and patient and family’s choice. An informed discussion on benefits and side effects with the family is mandatory to ensure compliance with the treatment and the management of side effects. Whichever steroid and regime is prescribed, the potential risk of adrenal failure should be considered when the child is unwell, in case of surgery and prior to anaesthetics. Each patient should be provided with an up-to-date alert card which details steroid treatment (including regime) and offers advice with regards to steroid replacement in emergencies.
Clinical Care Recommendations for Duchenne Muscular Dystrophy
As established by the care recommendations published in 2010, DMD is a complex multi-system disorder and requires a multidisciplinary approach. This involves different specialities such as neuromuscular specialists, paediatrics, cardiology, respiratory, orthopaedics, endocrinology, physiotherapy, occupational therapy, speech and language therapy, and psychology.2,3 The role of each speciality team varies according to the disease stage the patient is in and the progression of the symptoms, however all teams should be involved from the point of diagnosis. The role of the tertiary neuromuscular team is co-ordinating the patient care and ensuring that the right specialists are involved at the correct time to provide a proactive model of clinical care in which interventions (such as cardiac medications, access to non-invasive ventilation, etc.) can be swiftly organised when needed. Surveillance in DMD is centred around early detection of disease complications but also ensuring that patient-specific needs are identified and managed in order to optimise their physical, social, educational, and psychological status. The best practice of care in DMD has been translated into 30 different languages and a summary outlining the imperatives in DMD care has recently been published36 to facilitate optimal care to families living with DMD worldwide (Table 2). An update of the care recommendations is currently in progress reflecting the more recent changes and improvements in patient management.
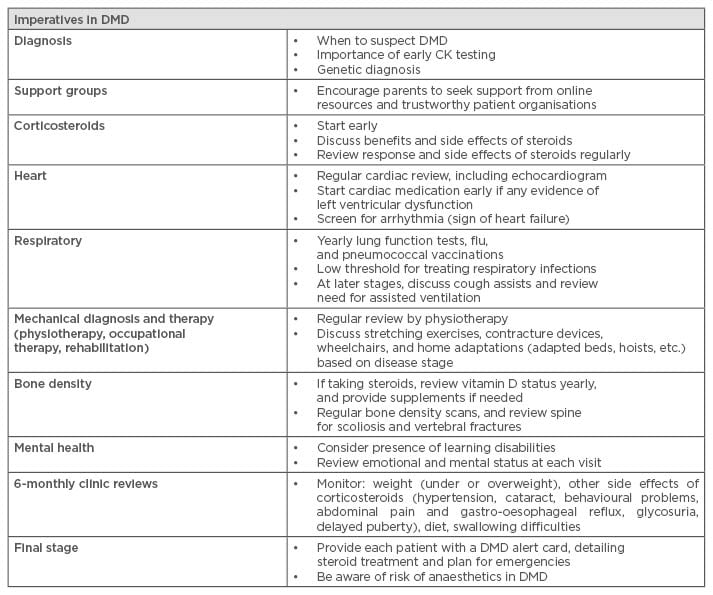
Table 2: Imperatives in Duchenne muscular dystrophy care.
DMD: Duchenne muscular dystrophy, CK: creatine kinase.
Regardless of the extensive effort in disseminating the DMD care recommendations worldwide, a recent survey has shown an unexpected discrepancy between the published recommendations and care provision as reported by patients and families across different countries.37 The survey highlighted concerns in all countries regarding access to routine follow-up by neuromuscular, cardiac, and respiratory specialists, physiotherapy, and access to medical devices. In addition, the survey showed a worrying lack of access to psychosocial therapy in DMD: this is of particular relevance in view of the continuous improvement in life expectancy for this patient population, leading to a new adult group of patients who live with a chronic and disabling condition and for whom psychological and social support will become essential to ensure good quality of life.
PROGNOSIS
Life expectancy for patients with DMD has increased dramatically over the last 20 years, with a mean age of survival currently between 23.0–27.8 years.38-41 The prolonged survival is the result of improved disease management leading to a delay in the development of complications. This includes clinical care, corticosteroid treatment, proactive cardiac monitoring and treatment, access to respiratory interventions, rehabilitation, and orthopaedic procedures. The main cause of death in DMD remains progressive cardiorespiratory failure, although the percentage of primary cardiac deaths in DMD is rising because management of respiratory complications has improved.42-44 Cardiac abnormalities in DMD progress with age and are characterised by initial left ventricular enlargement, followed by dilated cardiomyopathy with left ventricular dysfunction, arrhythmia, and heart failure.43 Whether prophylactic cardiac treatment is able to prevent or postpone the onset of cardiac involvement still needs to be demonstrated. Early onset cardiomyopathy has been associated with poor response to treatment, and worse prognosis.45
Early access to overnight ventilatory support has been the most successful intervention so far in prolonging survival in DMD.38 However, in practice this intervention is not always well-tolerated and therefore may be refused or delayed, increasing the risk of severe respiratory complications and death. Respiratory physiotherapy and proactive management of chest infection with up-to-date vaccinations, low threshold for antibiotic treatment, and devices to assist cough (e.g. Ambu bag, cough assist machine) can also impact outcomes.
Even with this positive trend and the continuous increase in survival rates in DMD, unexpected young deaths have been anecdotally reported. Teenage years seem to be a particularly fragile phase in this population and increased surveillance might be required in patients showing an unexpected deterioration around this time. Although the causes of these young deaths often remain largely unclear, several risk factors have been suggested. These include insidious worsening of respiratory function, presence of malnutrition, subtle cardiac arrhythmia, stress-induced adrenal failure in patients undergoing long-term corticosteroids treatment, fat embolism following trauma, psychological distress, and poor attendance to follow-up appointments. Awareness of these risk factors is important in order to minimise the risk of an untimely (sudden) death in DMD.46
THE FUTURE OF DUCHENNE MUSCULAR DYSTROPHY
The last 10 years have seen an enormous effort in developing new therapeutic approaches for DMD. Improvements in clinical care have already significantly changed the natural course of the disease, and recent investments in new potential treatments for the condition mean a promising future lies ahead. Several clinical trials in DMD are currently running, offering different approaches. These range from mutation-specific strategies aiming to correct the underlying genetic cause of the disease (exon skipping, reading through), to gene and stem cell therapy and non-mutation-dependent strategies directed to address downstream pathological pathways (anti-myostatin, utrophin, vamorolone, etc.).47 One of these approaches, tested in clinical trials, has led to the successful approval of a drug for DMD by the European Medicines Agency (EMA) called ataluren (Translarna™, conditional approval). Moreover, parallel studies investigating new treatments for the cardiac and respiratory complications (e.g. idebenone, eplerenone, nebivolol, etc.) have been developed in recent years. This illustrates that the clinical multidisciplinary approach also extends into the field of research. A list of the currently open studies is summarised in Table 3, however the list will continue to evolve over time.
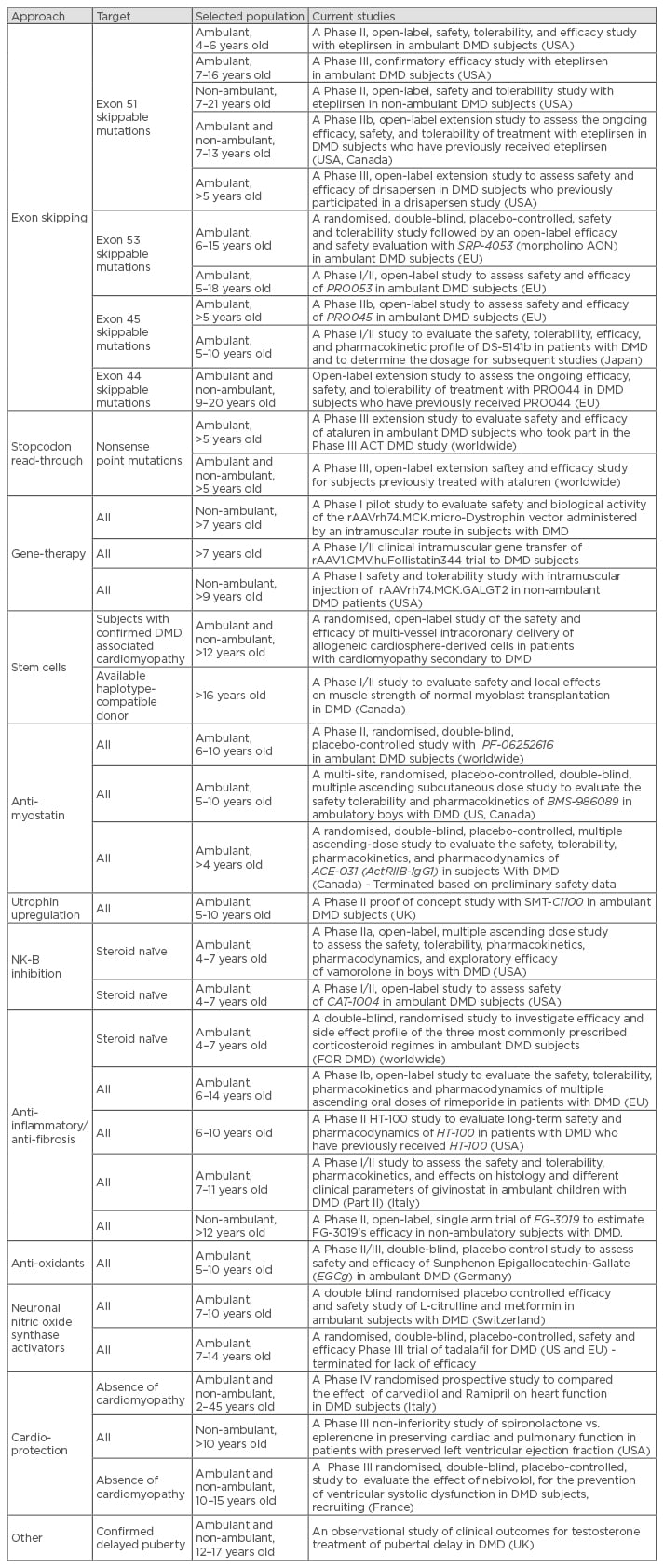
Studies non-specific for DMD (e.g. also recruiting patients with other neuromuscular conditions) are not included. Completed studies are not listed with the exception of two studies which were terminated only recently. Due to the continuous progress of the research in the field, this list is likely to change quickly and should therefore only be used as an indicator or number of studies in DMD and current approaches.
Full details of clinical trials in DMD can be found on www.clinicaltrials.gov
DMD: Duchenne muscular dystrophy; AON: antisense oligonucleotide; NK-B: neurokinin-B.
The rapid development of research in DMD has raised concerns on feasibility of multiple clinical trials in rare diseases where the patient population is limited, especially for targeted approaches (e.g. mutation-specific interventions). This is further complicated by the current requirements of a very narrow patient population (with regard to eligibility criteria; e.g. age, stage of the disease, mutation, etc.) being recruited in clinical trials, in order to reach study primary outcomes timely and efficiently. Currently a number of clinical trials exclude participants who have previously received any investigational drug, further obstructing recruitment and drug development in rare diseases. Although the results of some of the recent studies have been disappointing, lessons have been learnt from the clinical trial experience with the disease over the past years. This has raised awareness of the gaps which need to be addressed in order to ensure more cost-effective studies in the future and the ability to provide clearer outcomes on safety and efficacy of new investigational drugs. This includes: the importance of patient registries to allow identification of patients for large international studies; a better understanding of outcome measures for DMD; the impact of age, corticosteroid treatment, provision of standardised care on motor performance, and outcome measures. Other factors to take into consideration include the importance of disease-specific biomarkers breaching clinical outcomes; the possible impact of mutation type and location on clinical severity; the ethical requirement to involve older patients in clinical research, and the current lack of longitudinal natural history outcome data on this non-ambulatory population; the need for harmonisation of the regulatory processes, and requirements to facilitate the conduct of international studies in rare diseases. Finally, the need to develop a new health economic model applicable to rare diseases, and to ensure objective evaluation of the clinical effectiveness of investigational drugs without discouraging future drug development.48
CONCLUSION
Historically, DMD was considered an incurable condition and as such was approached with very limited proactive management. The development and dissemination of care recommendations for DMD has significantly changed this approach, leading to improved survival and quality of life, and allowing the prospect of an adult life for most patients diagnosed today. The field has seen large investments in terms of new therapeutic approaches which have now been taken into clinical trials and have resulted in one of the tested drugs receiving conditional approval by the EMA. Finally, there is hope that the future for boys and young men with DMD will significantly improve in the upcoming years. This leads to a new set of challenges, including social and psychological impacts of longer-term survival which need to be addressed in parallel with the continued improvement in care and treatment. The experience built over the last 20 years in DMD (implementation of diagnosis, dissemination of care recommendations, acquiring essential natural history data, and delivery of clinical research with investigational drugs) is likely to be of extreme value for the future of DMD patients. It also provides a learning experience for the development of new therapeutic approaches in other rare diseases.