Abstract
Guillain–Barré syndrome (GBS) is a form of rapidly progressive, acute inflammatory demyelinating polyradiculopathy. Acute inflammatory demyelinating polyradiculopathy is the most common variant of GBS, especially in the western hemisphere. It is diagnosed without hesitation when it presents with its characteristic clinical features of ascending paralysis and areflexia. It is when an atypical presentation appears, with brisk or very brisk reflexes, that diagnosis becomes difficult. In this case, a patient who presented with progressive motor weakness, unilateral facial palsy, and hyperreflexia on examination is described in order to demonstrate a variant of GBS and its management. Keeping in mind that the management of the disease does not change with the variant, the diagnostic challenge that is put forward by those variants needs a focussed approach by physicians.
INTRODUCTION
Guillain–Barré syndrome (GBS) is a form of rapidly progressive, acute inflammatory demyelinating polyradiculopathy (AIDP). AIDP is the most common variant of GBS, especially in the western hemisphere.1 This disease appears to be triggered by an infection or vaccination which disrupts self-tolerance, thereby leading to an autoimmune response which clinically presents as progressive motor weakness of one or more limbs with additional areflexia.2 Facial paresis in GBS is another common clinical finding, which is mostly bilateral; however, a rare variant of unilateral facial paresis can also occur.3 Sensory changes are also a common manifestation of most GBS variants. Bickerstaff’s brainstem encephalitis, a rare postinfectious inflammatory disorder, also shares overlapping features of similar aetiology with GBS, and together with Miller Fisher syndrome, they are considered as variables of the same neurological disease spectrum.4
This case represents a rare variance of GBS in a young male who presented with weakness in all four limbs with hyperreflexia and a unilateral facial palsy. The hyperreflexia is another clinically variant symptom of GBS that can produce a diagnostic dilemma in a patient with acute paresis. While most clinicians now accept that clinical variants, although rare, should be thoroughly investigated and pursued in the same manner as a usual case of GBS, on occasion a diagnostic dilemma can delay the management and worsen the prognosis with late intervention. In previous case studies there has been an argument to not expose the patients to immunotherapy in mild disease until there is absolute surety regarding the clinical diagnosis.5,6 The counterargument is that the delay in intervention can worsen the prognosis; there are multiple cases discussed below that show benefits of early therapy, with complete resolution of symptoms.7
CASE PRESENTATION
A 16-year-old male with no known comorbidities presented with complaints of weakness in all four limbs for the previous 9 days and pain in the left shoulder for 1 day. The weakness was sudden in onset and started in the lower limbs, with the patient suddenly falling while walking. He required assistance when standing back up, as well as when trying to walk. The weakness progressed the following day, which resulted in him being unable to get out of his bed or to perform daily chores. The weakness then spread to his upper limb and the patient said that he could not change his clothes without assistance. The patient also developed pain in the left shoulder for 1 day which was muscular in character. The pain was sudden in onset and increased with attempts at activity and decreased with rest. In the 6 days prior to the onset of weakness, the patient also had a history of fever which resolved on its own. However, this fever did not coincide with the muscle weakness of acute presentation. There was also a history of fever and pustular eruptions on the hands and feet 2 months prior, which were itchy and likely to be lesions from scabies. The lesions were resolved after the use of over-the-counter emollient creams. In the following months, the patient had repeated hospital admissions for pain crises, abdominal pain, diarrhoea, and weight loss. For every admission he was treated with red blood cell exchange transfusions, intravenous hydration, antibiotics, and analgesics; haemaglobin S levels were maintained at <28%. A lactose-free diet was recommended, but the patient continued to have six to eight loose stools per day. A flexible rectosigmoidoscopy revealed areas of exudate and necrosis in the rectosigmoid colon with multiple blood clots, and biopsies obtained did not show any significant findings. Stool cultures and stool parasitology tests were negative. Abdominal T2-weighted MRI to assess iron overload showed no signs of secondary liver haemochromatosis, with liver iron being 1.1 mg/g (dry liver measurement), but significant liver hypertrophy and upper abdominal varices were suggestive of portal hypertension. CT evaluation of the abdomen revealed bowel distention, thickening of the intestinal wall, mesenteric lymph node enlargement, signs of liver cirrhosis, and presence of ascites. Liver elastography showed an elevated liver stiffness of 13.2 kPa, indicative of liver cirrhosis.
On initial examination, the patient’s vital signs including body temperature, pulse rate, respiratory rate, and blood pressure were all in the normal ranges. Motor system examination revealed the muscle bulk and tone to be normal in all four limbs. For both the upper limbs, the grade of power was 2/5 in both the proximal and distal muscle groups. For the lower right limb, the grade of power was 2/5. For the lower-left limb, the grade of power was 4/5, which later decreased to 2/5 in both the proximal and distal muscle groups during another day of hospital stay. The deep tendon reflexes were brisk (+3) throughout all four limbs except for the patellar tendon reflex which had a very brisk response (+4). However, the plantar response was flexor, Hoffman’s sign was negative in the upper limbs, and abdominal reflexes were normal. The cranial examination revealed left-sided lower motor neuron facial palsy, as well as an absent corneal reflex, while the rest of the cranial nerve examination was unremarkable. The involvement of lower motor neurons in GBS is probably a result of infectious polyneuritis, which might involve or spare isolated cranial nerves. There was a blink response on the right with left corneal stimulation, ruling out the involvement of the V1 branch of the left trigeminal in addition to the left seventh cranial nerve. Other sensory modalities such as fine and crude sensations, two-point discrimination, pain, temperature, vibration, and proprioception were all intact. Examination of the shoulder joint revealed muscular spasm, but other signs of tendon injury or rotator cuff pathology were negative. Other systemic examination segments revealed no abnormalities. The differential diagnosis at this point was acute myelopathy (transverse myelitis), neurotoxic polyneuropathy (botulism, fish poisoning), spinal cord syndromes (postinfectious), toxic or vasculitic neuropathies, AIDP, and metabolic myopathies.
The laboratory workup findings were a haemoglobin count of 15.2 g/dL (normal 13.2–17.5 g/dL), erythrocyte sedimentation rate of 17 mm/hour (normal 0–15 mm/hour), total leukocyte count of 6.4×103/µL (normal 4.1–10.9×103/µL), platelet count of 342×103/µL (normal 150–400×103/µL), urea level of 32.3 mg/dL (normal 15.0–43.0 mg/dL), creatinine level of 0.62 mg/dL (normal 0.60–1.20 mg/dL), sodium level of 139 mmol/L (normal 134–144 mmol/L), chloride level of 100 mmol/L (normal 98–107 mmol/L), potassium level of 3.9 mmol/L (normal 3.6–5.0 mmol/L), bicarbonate level of 30 mmol/L (normal 23–30 mmol/L), total bilirubin of 0.34 mg/dL (normal 0.30–1.00 mg/dL), alanine aminotransferase level of 25 IU/L (normal 10–35 IU/L), aspartate aminotransferase level of 24 IU/L (normal <35 IU/L), alkaline phosphatase level of 154 IU/L (normal 44–147 IU/L), and γ-glutamyl transferase level of 18 IU/L (normal 15–80 IU/L). Viral serologies were negative for hepatitis, HIV, cytomegalovirus, Epstein–Barr virus, herpes simplex virus, and varicella-zoster virus. The West Nile virus and Lyme disease diagnostic tests were not available in the authors’ region and therefore were not performed.
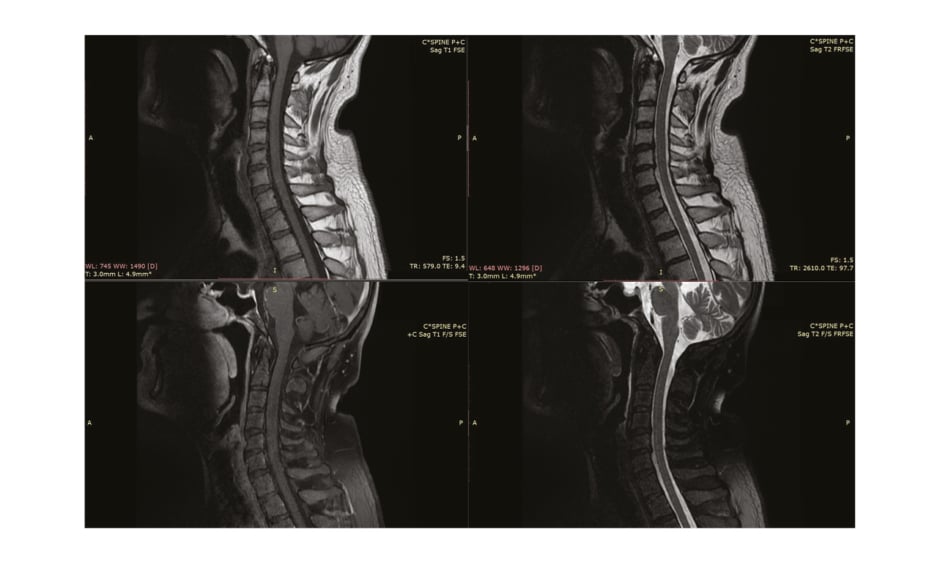
Nerve conduction studies performed in the initial week of symptoms onset were nonspecific to postinfectious pathology and revealed C8-T1 radiculopathy (Table 1). Needle electromyography was not performed in the patient. The lumbar puncture, preceded by a plain CT scan of the brain, revealed no findings and ruled out central nervous system inflammation (Figure 1). MRI (with contrast) of the cervical spinal cord was normal (Figure 2). Cerebrospinal fluid (CSF) examination on Day 10 of clinical onset revealed a cell count of 6 cells/mm3 (normal 100–1,000 cells/mm3), CSF protein level of 142 mg/dL (normal 15–40 mg/dL), CSF glucose level of 68 mg/dL (normal 40–70 mg/dL), CSF chloride level of 117 mEq/L (122–132 mEq/L), CSF white blood cell count of 0.001×103/µL (normal 0.000–0.005 103/µL), and no red blood cells or pus cells were detected, and hence was suggestive of albuminocytological dissociation.
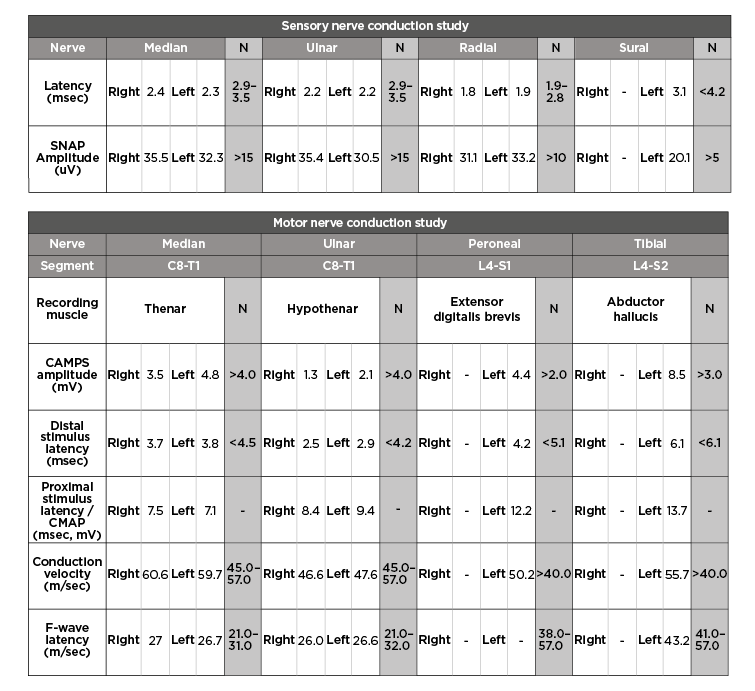
Table 1: Motor and sensory nerve conduction studies.
N: normal.
Figure 1: CT scan of the patient’s brain (without contrast, showing normal findings).
Figure 2: MRI of the patient’s cervical spine (with contrast, showing normal findings).
No other features in the history and examination were suggestive of toxic ingestion or autoimmune illness, hence the diagnosis of the patient was established to be GBS, and prompt action was taken to begin the patient’s treatment with one cycle of plasmapheresis every alternate day (40–50 mL/kg). The plasmapheresis was preferred instead of intravenous Ig in this patient strictly because of cost-effectiveness. The patient started improving greatly after the plasmapheresis cycles were commenced, and all signs of neuropathy and cranial nerve deficit began to decrease and almost completely disappeared in the following 2 weeks after completing five cycles of plasmapheresis. The patient was discharged and followed-up in the ambulatory clinical setting 2 weeks later and had complete resolution of symptoms. Ethical approval was taken in this study from the institutional review board, and consent to participate has been taken from the patient’s guardian with informed verbal consent.
DISCUSSION
GBS is a group of syndromes that are classified into demyelinating and axonal forms based on their pathologies. Approximately 1.72 patients per 100,000 people per year are considered to be affected by GBS, with a further increase of 50% with every 10-year increase in age.1 Despite this, it should be noted that although one of the fundamental features of GBS is hyporeflexia or areflexia, a patient presenting with preserved reflexes or hyperreflexia does not rule out GBS as a diagnosis.1 Hyperreflexia is known to be seen in patients with a previous Campylobacter jejuni infection with prior history of abdominal pain and diarrhoea; however, the most common and repeatable results have been seen with the acute motor axonal neuropathy type of GBS, which is seen in approximately 48% of Chinese patients and 33% of Japanese patients.2
It is important to note that although hyperreflexia is known to be seen in patients with previous C. jejuni infections, the patient in this present case had no previous history of such infection and the onset of the disease was more unforeseen, presenting with sudden onset of lower-limb paralysis that progressively worsened to all involve all four limbs.
This presentation shows that even though C. jejuni infection may show some correlation with hyperreflexia in patients, this rare presentation does not require a prior infection.2
The clinical course of GBS is usually aggressive and tends to involve respiratory muscles which may result in ventilatory failure and requirement of respiratory support in up to one-third of patients.2 Most patients also complain of sensory disturbances such as paraesthesia and numbness.
Although the presentation of hyperreflexia in GBS is not common but known,8 it is highly likely that it may be an associated finding in upper motor neuron dysfunction.
A common theory was that the role of anti-GM1 antibodies could cause the hyperreflexia by damaging nerve roots and central axons of the spinal cord when entering the subarachnoid space through the blood–brain and blood–spinal cord barriers.9
However, the exact mechanism of hyperreflexia in GBS and why nerve root damage does not cause hyporeflexia or areflexia, even if there is upper motor neuron dysfunction, is still unknown but is postulated to be associated with spinal inhibitor intermediate neuronal dysfunction.9 Few studies have shown that although there is a high incidence of antiganglioside (GM-1) antibodies being present in patients with hyperreflexia, it is still possible for patients to present with this rare variant without any antibodies present in their serum.3 Most patients with GBS show increased protein counts in the CSF, but nerve conduction studies remain the most sensitive diagnostic tool. However, electrodiagnostic studies cannot be highly sensitive in diagnosing GBS in the early course of the disease, as mentioned by several studies.10,11 The usual sensitivities to diagnose GBS with nerve conduction studies are 55–59%, according to two different studies.10,11
In literature, similar cases have been reported, and few of those discussed the need to withhold the immunotherapy till the second week to be sure of the diagnosis.5,6 Another review of two patients with facial palsy and hyperreflexia preceding C. jejuni infection and the presence of antiganglioside antibodies showed improvement with the administration of intravenous Ig.7 Intensive physiotherapy can help to reach complete resolution within 2 weeks.12 The role of intravenous methylprednisolone along with intravenous Ig was also discussed in one case.13 Hyperreflexia can persist, with complete resolution of muscle strength potentially taking up to 1 year.14,15 Follow-up nerve conduction studies usually show regenerative changes with the resolution of symptoms.16,17 The paediatric population usually presents with such a variant type of GBS,18,21 and it has been reported in women who are pregnant, but this is rare.22 Asian and European populations are more commonly affected by this variant.2,23 Bilateral facial palsy is rarely the only presentation of this variant;24 although, almost one-third to one-half of the patients with GBS have facial palsy, either unilateral or bilateral.14,15,25
Hemiplegia and cranial nerve palsies are the other reported variants of GBS that are closely related.26 The overall prevalence of this GBS variant is reported to be around 0.2–0.8%;14,15,23,24 however, hyperreflexia can be present in 33–48% of AIDP cases.20,26 Miller Fisher syndrome is another closely related variant, which can involve extraocular muscles and diminished reflexes, in contrast to this present case.27 Bickerstaff’s brainstem encephalitis, which consists of a triad of ophthalmoplegia, ataxia, and altered sensorium, is another closely related disorder that shares overlapping features and similar aetiology to GBS. The only feature that differentiates the two entities is altered level of consciousness, which was absent in the author’s case.4,28 Recent Zika virus outbreaks were also reported to be associated with GBS, hyperreflexia, and facial or bulbar palsy, but myelopathic forms appeared with different prognoses and high mortality.29 The development of hyperreflexia at the knees and normoreflexia at the ankles in a teenage male can be attributed to many other causes, including medication and stimulant side effects, hyperthyroidism, electrolyte imbalance, serotonin syndrome, severe brain trauma, and Reye’s syndrome. A specific cause has been recently identified as L5 nerve root injury.30
CONCLUSION
This case report shows that although areflexia and progressive motor weakness are both key elements of GBS, if a patient does not present with both, GBS should still be in the physician’s working differentials. The aim of this case report is to increase the awareness regarding this different variant of GBS, which was not diagnosed with nerve conduction studies but by meeting other criteria. Although it is rare, prompt diagnosis and management showed worthiness in improving prognosis in various case studies.