Meeting Summary
The maintenance of mitochondrial DNA (mtDNA) is dependent upon several nuclear gene-encoded proteins including enzymes forming the replisome needed to synthesise mtDNA. These enzymes need to be present in balanced quantities to function properly. In addition, mtDNA synthesis requires a balanced supply of nucleotides that is achieved by nucleoside recycling inside the mitochondria, and nucleotide import from the cytosol. Mitochondrial DNA maintenance defects are a group of diseases caused by pathogenic variants in the nuclear genes involved in mtDNA maintenance, and result from impaired mtDNA replication. Pathogenic nuclear gene variants identified to date include genes that encode enzymes of mtDNA replication machinery (such as POLG), genes that encode proteins that help to maintain a balanced mitochondrial nucleotide pool (such as TK2), and genes that encode proteins involved in mitochondrial fusion. Here, the presentation provided by Ramon Martí, Research Group on Neuromuscular and Mitochondrial Diseases, Vall d’Hebron Institut de Recerca, Barcelona, Spain, and CIBERER, Madrid, Spain, is summarised. A leading expert on mitochondrial pathology, Martí presented at the Euromit 2023 International Conference on Mitochondrial Disease, which took place in Bologna, Italy, in May 2023.
Introduction
Mitochondria are organelles present in almost all eukaryotic cells. Responsible for orchestrating cellular energy production, they are central to the maintenance of life and the gatekeepers of cell death. Many mitochondrial diseases originate from pathogenic mtDNA mutations (Figure 1)1 that lead to defects in various mitochondrial proteins, disrupting the electron transport chain and oxidative phosphorylation (OXPHOS). With these dysfunctions, mitochondria are unable to produce sufficient energy in different tissues, especially in the highly adenosine triphosphate-demanding tissues, such as skeletal muscle, cardiac muscle, liver, and the renal and central nervous systems.2,3 An increasing number of mutations in nuclear genes involved in deoxyribonucleotide homeostasis have been identified in causing disorders associated with somatic mtDNA abnormalities. Dysfunction of the products of these genes causes limited availability of substrates for mtDNA replication, which leads to multiple deletions, point mutations, or mtDNA depletion. The latter is the molecular feature linked to greatest clinical severity.3,4 At the Euromit 2023 International Conference, Martí presented recent results demonstrating that enhancement of the salvage pathways by increasing the availability of deoxyribonucleosides improves the mtDNA copy number. Martí further discussed the administration of selected deoxyribonucleosides as a potential pharmacological strategy to treat mtDNA depletion/deletions syndromes (MDDS).1
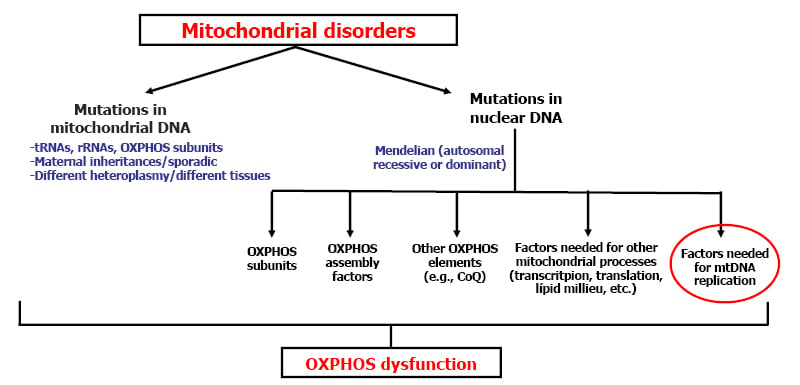
Figure 1: Causes of mitochondrial disorders.1
CoQ: co-enzyme Q10; mtDNA: mitochondrial DNA; OXPHOS: oxidative phosphorylation; rRNA: ribosomal RNA; tRNA: transfer RNA.
Mitochondrial Diseases
Martí opened their talk by explaining that mtDNA contains genes necessary for mitochondrial OXPHOS function. The mitochondrial OXPHOS system is the final biochemical pathway, whereby cells use carbon fuels and O2 to produce adenosine triphosphate.5,6 In addition, many nuclear encoding proteins are needed for the function of the OXPHOS system. Human mtDNA is a double-stranded, circular molecule of 16.6 kb, and contains 37 genes, which encode for two ribosomal RNAs, 22 mitochondrial transfer RNAs, and 13 polypeptides involved in OXPHOS.6,7 Many mitochondrial diseases are caused by mutations in mtDNA, but also by mutations in nuclear DNA encoding different factors, including those needed for mtDNA replication (Figure 1).1
When mtDNA replication is dysfunctional due to gene defects, there can be a reduction in the quality of the mtDNA due to multiple deletions and point mutations, or reduction in quantity of mtDNA (depletion) in affected tissues.8,9 Disorders of mtDNA maintenance are clinically heterogeneous and severe due to mtDNA depletion and multiple deletions.8 Mitochondrial DNA is replicated and repaired by nuclear-encoded mtDNA POLG and several other associated proteins, which compose the mtDNA replication machinery.10 To achieve correct replication of mtDNA, Martí explained that functional machinery is needed, but also a correct supply of deoxynucleoside triphosphates (dNTP), the building blocks of new DNA strands made during DNA replication.9,11 A central enzyme for generating dNTPs is ribonucleotide reductase.9 Martí then reviewed the genes identified so far to be associated with mtDNA maintenance disorders, including a group of genes (Figure 2) that encode not only mitochondrial proteins but also cytosolic proteins that participate in dNTP homeostasis.9
Currently, five key nuclear genes encoding proteins that participate in dNTP homeostasis have been identified to be associated with MDDS. These genes are TYMP, TK2, DGUOK, and RRM1 and RRM2B (encoding subunits of the ribonucleotide reductase).9,12 Mutations in any of these nuclear genes can lead to imbalanced dNTPs and impaired mtDNA replication.4,13,14
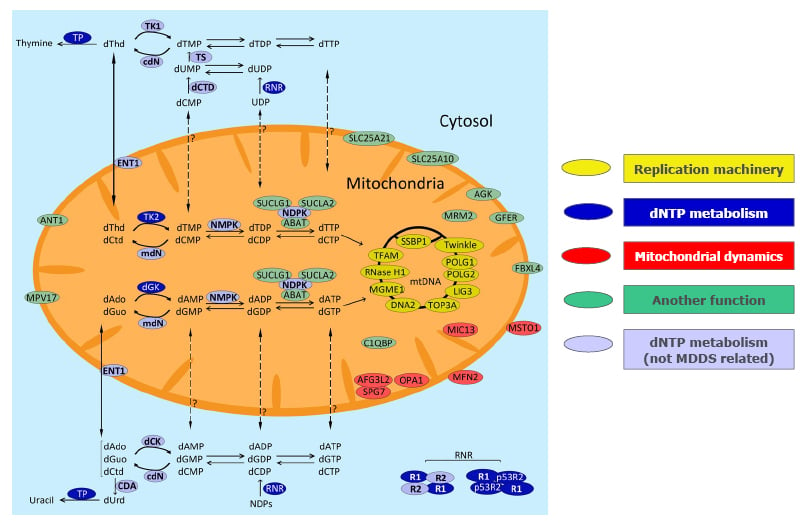
Figure 2: Genes (proteins) associated with mitochondrial DNA maintenance disorders.9
Proteins whose mutations have been linked to MDDS are depicted in yellow (mtDNA replication/maintenance machinery), dark blue (dNTP metabolism), red (mitochondrial dynamics), and green (unknown role in mtDNA replication). Light blue colour represents proteins participating in dNTP metabolism but not associated with MDDS.
Modified from Ramón et al.9
ABAT: 4-aminobutyrate aminotransferase; AFG3L2: AFG3-like protein 2; AGK: acylglycerol kinase; ANT1: adenine nucleotide translocator 1; CDA: cytidine deaminase; cdN: cytosolic deoxyribonucleotidase; C1QBP: complement component 1 Q subcomponent-binding protein; dAdo: deoxyadenosine; dCK: deoxycytidine kinase; dCtd: deoxycytidine; dCTD: deoxycytidylate deaminase; dGK: deoxyguanosine kinase; dGuo: deoxyguanosine; DNA2: helicase/nuclease DNA2; dNTP: deoxynucleotide triphosphate; dThd: deoxythymidine; dUrd: deoxyuridine; ENT1: equilibrative nucleoside transporter 1; FBXL4: F-box/LRR-repeat protein 4; GFER: growth factor, augmenter of liver regeneration; LIG3: ligase III; MDDS: mitochondrial DNA depletion/deletions syndromes; mdN: mitochondrial deoxyribonucleotidase; MFN2: mitofusin 2; MGME1: mitochondrial genome maintenance exonuclease 1; MICOS13: MICOS complex subunit MIC13; MPV17: protein MPV17; MRM2: rRNA methyltransferase 2; MSTO1: protein misato homolog 1; mtDNA: mitochondrial DNA; NDPK: nucleoside diphosphate kinase; NMPK: nucleoside monophosphate kinase; OPA1: dynamin-like 120 kDa protein; POLG1: catalytic subunit of polymerase gamma; POLG2: ancillary subunit of polymerase gamma; p53R2: p53-inducible small subunit of the ribonucleotide reductase; R2: small subunit of the ribonucleotide reductase; RNASEH1: ribonuclease H1; RNR: ribonucleotide reductase; SLC25A10: mitochondrial dicarboxylate carrier; SLC25A21: mitochondrial 2-oxodicarboxylate carrier; SPG7: paraplegin; SSBP1: mitochondrial single strand binding protein; SUCLA2: subunit of the succinate-co-enzyme A ligase; SUCLG1: subunit of the succinate-co-enzyme A ligase; TFAM: mitochondrial transcription factor 1; TK1: thymidine kinase 1; TK2: thymidine kinase 2; TOP3A: DNA topoisomerase 3-alpha; TP: thymidine phosphorylase; TS: thymidylate synthase; Twinkle: mitochondrial helicase.
Mitochondrial DNA Replication
Martí studied the replication of mtDNA in isolated mitochondria using an experimental murine liver model, and showed that an excess of deoxythymidine triphosphate (dTTP) decreased mtDNA synthesis, but this effect was due to secondary deoxycytidine triphosphate (dCTP) depletion rather than the excess dTTP.15 The group found that the addition of dCTP restored mtDNA replication in the presence of thymidine overload, thus confirming that thymidine excess in itself does not slow down mtDNA replication. Their group showed that by supplementing dTTP excess with an excess of dCTP caused the recovery of mtDNA levels, indicating that the availability of dNTP is the key factor that leads to mtDNA depletion.15 This was confirmed in human cultured cells. Martí discussed that the delay of mtDNA replication rate observed when dTTP is in excess produces a TK2-mediated secondary dCTP depletion that actually delays mtDNA replication. Therefore, the common factor accounting for mtDNA depletion in these disorders is the limited availability of one or more nucleotides.15 Martí concluded that, as far as we know, all the mtDNA replication disorders directly related to disturbed dNTP homeostasis are, in fact, caused by limited availability of one or more dNTPs.1 This study indicated that strategies to provide nucleotides to patients’ mitochondria should be explored as a possible treatment for these fatal disorders.15
TK2 Deficiency
Martí went on to discuss TK2, a nuclear-encoded mitochondrial enzyme that catalyses the conversion of deoxythymidine and deoxycytidine nucleosides to their nucleoside monophosphates that, in turn, are further phosphorylated to generate dTTP and dCTP, which are incorporated into replicating mtDNA.9,16 TK2 deficiency can present as various phenotypes; the predominant one is moderate-to-severe myopathy.9 Patients with infantile onset are most severely affected, presenting with severe myopathy and only ~25% survival 2 years post-diagnosis.16
Treatment Option for TK2 Deficiency
Martí then explained that the first biochemical strategy to treat TK2 deficiency was the administration of thymidine and deoxycytidine monophosphates (nucleotides) to patients to correct this mitochondrial defect.1 Martí reviewed that the efficacy of this approach was actually demonstrated in 2014 by Garone et al.,17 in which they orally supplemented a TK2 deficient animal model (a TK2 knock-in mouse) with deoxycytidine and deoxythymidine monophosphates. The group found that untreated animals lived for approximately 13 days; however, if they received monophosphates, they lived longer, in a dose-dependent manner with a response to treatment with these compounds. In these mutant animals, the addition of monophosphate compounds raised dTTP concentrations, increased levels of mtDNA, ameliorated defects of mitochondrial respiratory chain enzymes, and significantly prolonged their lifespan (34 days with treatment versus 13 days untreated).17 Martí explained that this was the first demonstration that this supplementation could be a potential therapy for this disorder.
This raised some questions because the monophosphates are charged compounds that cannot pass across plasma membranes, so they cannot be absorbed as monophosphates through the intestinal epithelium, and even if you inject it into the blood, they will not enter the cells. However, these nucleotides are rapidly degraded by extracellular ectonucleotidases and phosphatases to the nucleoside, which is uncharged, and to the phosphate. Martí resolved that this enables the nucleoside to enter, because there are specific transporters that allow them entrance not only here but also to the mitochondria. So, at this point, their group thought that probably the effect was not by the monophosphates but by the nucleosides.1
Martí then raised the questions: “Why administer nucleosides if you still have this step blocked? Why would you need to do this?” Martí answered these by explaining that you are giving something that is before the blocking. They clarified that their group thought that these nucleosides are using their corresponding or their equivalent cytosolic enzymes that catalyse the same reactions but in the cytosol. The excess of substrate provides an extra amount of nucleosides to push the reaction towards the anabolic direction due to two complementary effects: saturation of the cytosolic salvage enzymes (or even some residual mitochondrial TK2 activity, if present) with excess of substrate increasing their catalytic rate, and the excess of substrates, in itself, displaces the reaction towards the reaction products (the nucleotides), due to the thermodynamic balance of the reaction.4,18 This enables nucleotides to enter mitochondria and restore normal mtDNA replication rate.
Martí discussed that Lopez-Gomez et al.19 and Blázquez-Bermejo et al.20 tested the nucleosides. Lopez-Gomez et al.19 tested the administration of deoxythymidine plus deoxycytidine, and the co-administration of the deaminase inhibitor, tetrahydrouridine, with thymidine monophosphate plus deoxycytidine monophosphate in TK2-deficient mice, and found that deoxycytidine plus deoxythymidine delayed disease onset, prolonged lifespan, and restored mtDNA copy number, as well as respiratory chain enzyme activities and levels. Blázquez-Bermejo et al.20 tested daily deoxythymidine plus deoxycytidylate deaminase (dCtd), thymidine monophosphate plus deoxycytidine monophosphate, deoxythymidine alone, or deoxycytidylate deaminase alone to TK2 knockout mice, and found that deoxythymidine plus deoxycytidylate deaminase treatment extended the average lifespan of TK2 knock out mice from 16 to 34 days, attenuated growth retardation, and rescued mtDNA depletion in skeletal muscle and other target tissues. These works indicated that the effect is produced by the nucleosides and not the monophosphates.19,20
POLG
POLG is the polymerase that replicates mtDNA.14 Mutations in POLG can cause early childhood mtDNA depletion syndromes, or later-onset syndromes arising from mtDNA deletions.9 POLG mutations are the most common cause of MDDS, producing a wide range of clinical presentations, some of which are severe, including Alpers–Huttenlocher syndrome, ataxia neuropathy spectrum, and progressive external ophthalmoplegia.9
Treatment Option for POLG-related Disorders
There are many known mutations in the POLG nuclear gene.21 Consequently, Martí’s group hypothesised that for those mutations that affect the affinity of the polymerase for the dNTPs, treatment with the nucleosides could also be effective. Martí explained that if there is a reduction of the affinity because of the mutation, and if you provide more dNTPs through the precursors that have the nucleosides, you could also counteract the problem. As there were no appropriate animal models, Martí’s group assessed their theory in quiescent fibroblasts isolated from patients harbouring mutations in different domains of POLG, and measured mtDNA copy number recovery rates following ethidium bromide-forced depletion. They found that when the ethidium bromide was withdrawn, the fibroblast controls recovered the amount of mtDNA. However, fibroblasts with mutations in POLG were not able to repopulate mtDNA copy number after ethidium bromide was withdrawn. Martí further showed that supplementation of the cells with nucleosides counteracts and prevents this mtDNA replication defect, regardless of the specific POLG mutation.14 Their group has repeatedly observed that nucleoside supplementation promotes mtDNA repopulation following this design, for all POLG pathogenic mutations tested.4,14 Martí emphasised that the important thing is that they had a positive effect for the whole range of POLG mutations, indicating that maybe, for some reason, this is not only working in mutations that affect their affinity of the polymerase for the dNTP, but may be useful for all mutations.
Martí continued that their group then hypothesised that the mechanism would be a different one. For enzymes following Michaelis–Menten kinetics, such as POLG, the reaction rate (velocity of the reaction catalysed by the enzyme) increases as the substrate concentration augments, following a known mathematical model (Michaelis–Menten equation). When you have saturating substrate concentrations, you reach the maximum velocity; however, if you have a low substrate concentration the rate of the reaction responds to increases of the concentration of the substrate. So, Martí’s group’s hypothesis was that if in normal conditions, you have a normal rate of replication, if you increase or you do an expansion of the entities by supplying nucleosides, you would accelerate polymerase, and then in this way you would counteract whatever defect the mitochondrial replication is having because you are forcing or pushing the mtDNA replication. So, you are probably incrementing or enhancing the synthetic pathway. But in order for this to be a possible sub-mechanism, there are two conditions that should be accomplished. One of them is that, in normal conditions, ie., in the conditions in which you do not treat the cells or the patients, the mitochondrial substrate (dNTP) concentration is not excessively above the Km (i.e., the enzyme is not saturated), so the reaction rate can respond to substrate concentration increases. Martí cited that the concentration of the substrate is similar to or around the Km (also known as the Michaelis constant), the substrate concentration at which the reaction rate is 50% of the Vmax or the maximum rate of the reaction. Km is a measure of the affinity an enzyme has for its substrate, as the lower the value of Km, the more efficient the enzyme is at carrying out its function at a lower substrate concentration) because if, in normal conditions for the tissue that you want to treat, you are at levels that are already saturated, you would not obtain a response. Martí emphasised that one condition is that in the normal or baseline situation that we want to correct, the concentration of the substrates of the dNTPs is low. The second condition is that when we treat the patient or the animals with deoxynucleosides, this significantly increases the concentration of the dNTPs because otherwise there would not be any effect.1,22
Martí explained that there is wide variability in experimental estimations of the Km for POLG for different nucleotides.23-31 Martí reviewed these estimations to select a Km of approximately 1 µM for their hypothesis. A higher Km would be easier to prove; therefore, this lower estimation would really challenge the hypotheses. Values for the dNTPs were provided by Vila-Julià et al.32 This study examined the mitochondrial brain dNTP in wild-type and mutant (knockout for TYMP and UPP1) mice, and the authors found that exposure of the animals to a continual dose of the nucleosides deoxythymidine and deoxyuridine in their drinking water resulted in a two-fold increase in mitochondrial dNTP levels.32 The exposed mice had more or less double the level of brain dTTP compared with the non-exposed ones. For the mutants, who at baseline already had double the level of dNTP, levels were further doubled after oral nucleoside administration. Martí emphasised that there is variability; however, their group was able to double the concentration of the dNTP in these conditions.32
For values in cells and tissues, Martí used a range between 1 and 10 μmol per mg mitochondrial protein. With this combination, Martí then estimated the dNTP concentration range between 1.3 and 13 µM. The experimental results are shown in Figure 3, along with a table obtained from Wendelsdorf et al.33 that shows different Km values depending on the status of the tissue. Martí stated that, “of course, these are probably levels of dNTPs of replicating cells that need more dNTPs.” However, these values are consistent. Combining the estimate of Km with the estimate of the concentration that was calculated, Martí was able to achieve the deoxynucleoside-induced dNTP expansion of mtDNA replication by saturating POLG.32,33
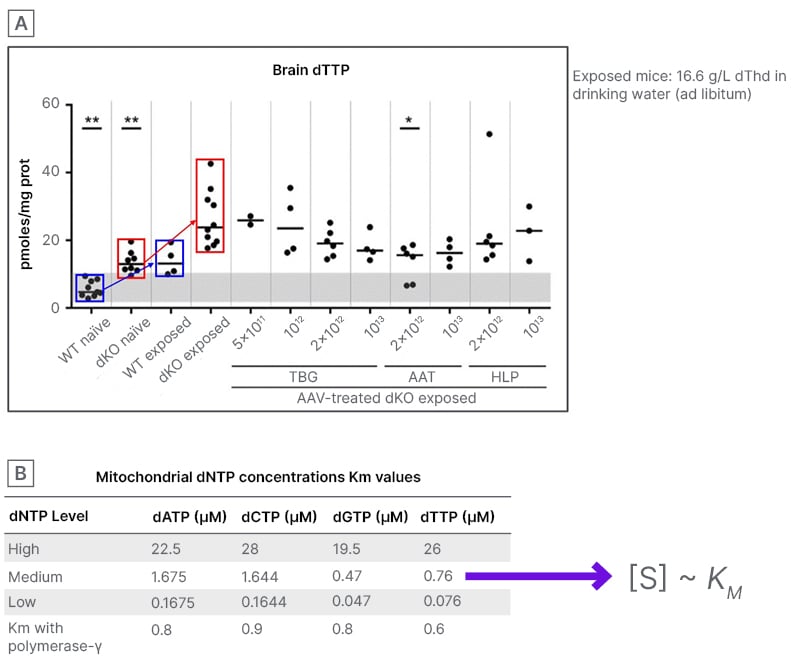
Figure 3: POLG enzyme kinetics data and deoxynucleoside triphosphate levels support the notion
that deoxynucleoside -induced deoxynucleoside triphosphate expansion enhances mitochondrial
DNA replication32,33
A) modified from Vila-Julià et al.;32 and B) modified from Wendelsdorf et al.33
AAT: α1-antitrypsin; AAV: adeno-associated virus; dATP: deoxyadenosine triphosphate; dCTP: deoxycytidine triphosphate; dGTP: deoxyguanosine triphosphate; dKO: double knockout; dNTP: deoxynucleoside triphosphates; dTTP: deoxythymidine-triphosphate; HLP: hybrid liver-specific promoter; TBG: thyroxine-binding globulin; WT: wild type.
Martí then explained that using the Michaelis–Menten equation, they were able to show that the sustained concentration of the dNTPs in the quiescent situation is more or less similar to Km, which means that the rate of the reaction is 50% of the maximum velocity. However, if the substrate concentration is doubled to 2 Km, then Martí was able to increase the velocity of reaction to 67% of the maximum. This 33% increase indicates an ability to significantly increase the rate of the semantic fate of the polymerase gamma in this condition.1
Martí concluded that the hypothesis demonstrated that this strategy of administering nucleosides can produce dNTP in an anabolic way, either mediated by the functional cytosolic enzymes or via the mitochondrial pathway. Martí stressed that in cases of mutations of POLG, there is also the mitochondrial pathway. This increase that is produced is stimulating the activity of POLG, increasing the rate of mtDNA replication. Martí’s group has also seen positive effects for this strategy in other diseases. Further investigation is needed to validate these results.1
Conclusion
MDDS constitute a group of genetic diseases defined by dysfunctional mtDNA replication and maintenance. Currently, the options for the treatment of these disorders are limited. Administration of selected deoxyribonucleosides may offer an expanded treatment armamentarium for patients with some forms of MDDS, such as TK2 deficiency and POLG-related disorders.