Abstract
Lung cancer is one of the most common malignant tumours worldwide, with non-small cell lung cancer (NSCLC) accounting for the largest number of cases among both men and women. Poor patient prognosis due to therapeutic resistance remains a current issue, underscoring the need for a more comprehensive understanding of the underlying biology of the pathogenesis and progression mechanisms of NSCLC. Integrating multi-omics approaches, such as genomics, transcriptomics, proteomics, and metabolomics, has become crucial for studying the underlying biology of complex diseases like lung cancer. Applying these methods not only enhances knowledge of the mechanisms of lung cancer but also plays a pivotal role in identifying biomarkers and therapeutic targets for implementing personalised treatment plans. This review quantitatively analyses the predictive capability of integrated multi-omics models by synthesising findings from studies utilising clinical data (including survival outcomes and treatment response) with multi-omics technologies to pinpoint essential biomarkers and pathways associated with NSCLC. The author focused on comparing the reported predictive accuracy metrics of these models and the consistency of identified key biomarkers across different studies. The author highlights the importance of integrating multi-omics analyses in the development of targeted therapies, and offers a roadmap for future clinical applications, emphasising challenges in data integration and biomarker validation, alongside opportunities for novel clinical trial designs. This review aims to provide a comprehensive quantitative assessment of the current state of integrated multi-omics in NSCLC, ultimately informing the design of more effective personalised therapeutic strategies and future research directions.
Key Points
1. Non-small lung cancer (NSCLC) accounts for approximately 85% of lung malignancies and remains the leading cause of cancer mortality worldwide, underscoring the urgent need for biomarker-driven precision strategies.2. This systematic review synthesised evidence from 50 original and peer reviewed research articles published during the last 5 years, integrating genomics, transcriptomics, proteomics, metabolomics, and epigenomics to evaluate biomarker discovery, patient stratification, and therapy optimisation in NSCLC.
3. Multi-omics integration enables clinically actionable biomarker identification, enhances prediction of immunotherapy response, and informs personalised treatment frameworks. Collaborative validation and translational pipelines are essential to embed these insights into routine NSCLC care.
INTRODUCTION
Lung cancer remains the most frequently diagnosed malignancy and the leading cause of cancer tumour-related mortality worldwide.1 Non-small cell lung cancer (NSCLC) is the primary subtype of lung cancer, accounting for 85% of lung cancer cases.2 Despite advances in targeted therapies and immunotherapies, therapeutic resistance remains the principal barrier to durable responses and long term survival.3,4 Resistance mechanisms span the genome, epigenome, transcriptome, proteome, metabolome, and tumour microenvironment (TME), and evolve dynamically during treatment. Understanding how these mechanisms interact across molecular layers is essential for improving treatment efficacy and patient outcomes.4
NSCLC arises from a multifactorial aetiology involving both environmental and intrinsic factors. Environmental exposures such as tobacco smoke, air pollution, and occupational carcinogen exposure induce chronic epithelial damage and mutagenesis, while internal factors, including genetic predisposition, immune dysregulation, and chronic inflammation, contribute to tumour initiation, progression, and immune escape. These aetiologic drivers vary in impact across histological subtypes and patient populations, underscoring the need for stratified prevention and therapy strategies.
Histologically, NSCLC is classified into three major subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (Supplementary Table 1). Each subtype exhibits distinct pathological characteristics, clinical behaviours, and molecular signatures. Biomarkers such as EGFR, ALK, and programmed death-ligand 1 (PD-L1) are routinely used to guide diagnosis and therapeutic decisions, particularly in the context of targeted therapy and immune checkpoint inhibition.5-7 However, their predictive power is limited by the emergence of acquired resistance, which often involves bypass signalling, epigenetic reprogramming, and TME-mediated immune evasion.
While blood-based biomarkers such as circulating proteins and cytokines offer a minimally invasive route for early detection and longitudinal monitoring, their clinical translation remains limited by challenges in specificity and validation across diverse populations. Expression levels of IL-6, IL-8, and colony stimulating factor 1 (CSF-1), for example, are influenced by systemic inflammation, comorbid conditions, and sampling variability, complicating their interpretive value in NSCLC.8,9 Similarly, epigenomic markers, particularly DNA methylation signatures, show promise for subtype classification and risk stratification. However, methylation patterns are highly context dependent, varying with tumour subtype, anatomical sites, environmental exposures, and immune status.10,11 These limitations underscore the need for multi-layered integration and robust validation frameworks, reinforcing the rationale for multi-omics approaches that triangulate signals across platforms to improve biomarker fidelity and clinical relevance.12-16
Recent concepts in tumour biology highlight how cancers remodel their microenvironment and hijack systemic homeostasis, including neuroendocrine signalling pathways that influence immune function, metabolism, and stress response.12 These interactions shape the natural history of NSCLC and complicate therapeutic design, reinforcing the need for narrative frameworks that capture tumour-intrinsic and host-mediated dynamics.
Multi-omics technologies offer a comprehensive approach in interrogating NSCLC biology that predicts therapeutic efficacy and for discovering personalised targets.13,14 The term ‘omics’ refers to a suite of technologies: genomics (DNA sequencing to identify actionable mutations), transcriptomics (RNA profiling reveals regulation and treatment response), proteomics (protein level analysis revealing tumour progression and resistance mechanisms), metabolomics (small metabolites profiling for therapeutic targeting and disease monitoring), and epigenomics (genome wide assessment of epigenetic regulation). The integration of these platforms enables the discovery of biomarkers that predict therapeutic efficacy, inform personalised treatment strategies, and support the development of precision oncology studies.13,15
The TME, a complex ecosystem of cancer cells, stromal components, immune infiltrates, and extracellular matrix, plays a pivotal role in NSCLC resistance progression. Single-cell multi-omics technologies have revealed profound TME heterogeneity and cell-type-specific drivers of resistance and progression, while systems biology approaches integrate multi-layer data to establish a causal link between molecular alterations and phenotypic outcomes.15-17 Proteomics studies, for example, connect genomic alterations with protein-level consequences, exposing therapeutic vulnerabilities that would be missed by single-platform analyses. Emerging evidence also implicates the microbiome as a modulator of host immunity and therapeutic response, particularly in immunotherapy.17,18 Metagenomic and whole-genome sequencing of microbial communities, including bacteria, fungi, viruses, and archaea, have revealed associations between microbial signatures and treatment outcomes.
Pharmacokinetic-pharmacodynamic modelling represents another frontier in NSCLC research. These mathematical frameworks simulate drug–tumour interactions, enabling the optimisation of radiotherapy and chemotherapy schedules and the prediction of resistant subclones, and guiding the design of regimens that minimise resistance and enhance efficacy.19,20
In summary, NSCLC exemplifies the complexity of cancer biology, where genetic, epigenetic, metabolic, and microenvironmental factors converge to drive resistance. Multi-omics integration provides the most promising avenue to unravel this complexity, enabling the development of biomarker-guided strategies that anticipate resistance, personalise therapy, and ultimately improve patient outcomes.
MULTI-OMICS INTEGRATION IN NON-SMALL CELL LUNG CANCER RESISTANCE
Investigating the intricate molecular changes that drive resistance to targeted therapies is crucial for understanding tumour cell survival and clinical progression during treatment. In recent years, therapies targeting EGFR, BRAF, and KRAS mutations and ALK, ROS-1, RET, NTRK fusions/rearrangements have improved survival in subsets of patients. Nevertheless, treatment responses remain incomplete, with acquired resistance emerging as the rule rather than the exception.21 Resistance mechanisms, including genetic mutations, epigenetic reprogramming, metabolic adaptation, and TME-mediated immune evasion, complicate the durability and the effectiveness of targeted and immune-based therapies (Supplementary Table 2). Therefore, multi-omics approaches, by interrogating tumour biology both at baseline and longitudinally during therapy, present promising tools to address these challenges.21,22
The interplay between genetic mutations and the TME is central to NSCLC complexity. Understanding these factors is vital for developing effective therapies and overcoming resistance in patients with NSCLC.
Emerging concepts further highlight how tumours regulate their environment and hijack systemic homeostasis through neuroendocrine signalling, influencing immune function, metabolism, and stress responses. These interactions shape the natural history of NSCLC and complicate therapeutic design, reinforcing the need for frameworks that capture both tumour-intrinsic and host-mediated dynamics.12
While blood-based biomarkers such as circulating proteins and cytokines offer a minimally invasive route for early detection and longitudinal monitoring, their clinical translations remain limited by challenges in specificity, reproducibility, and validation across diverse populations.
Expression levels of IL-6, IL-8, and CSF-1, for example, are influenced by systemic inflammation, comorbid conditions, and sampling variability, complicating their interpretive value in NSCLC.8,17 Similarly, epigenomic markers, particularly DNA methylation signatures, show promise for subtype classification and risk stratification. However, methylation patterns are highly context-dependent, varying with tumour subtype, anatomical site, environmental exposures, and immune status.23,11 These limitations underscore the need for multi-layered integration and robust validation frameworks, reinforcing the rationale for multi-omics approaches that triangulate signals across platforms to improve biomarker fidelity and clinical relevance.12,16
From DNA to RNA and ultimately to proteins, the amount and complexity of information progressively increases. Genomics has revealed key mutations, supporting the development of targeted therapies in personalised medicine.17 In NSCLC, genomic methods have identified aberrant activation of the PI3K/protein kinase B (AKT)/mTOR pathway as a driver of resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI). The discovery led to drugs targeting mTOR, EGFR, and ALK, which have shown clinical effectiveness in lung cancer treatment.24 However, the intricate nature of cancer mechanisms makes it difficult to establish clear links between tumours and specific genetic variants, necessitating a more nuanced approach to tackle its challenges.
Transcriptomics captures dynamic RNA profiles reflecting cellular states,17,25 while proteomics reveal functional protein changes, including post-translational modifications (phosphorylation, acetylation, and glycosylation) that change proteins’ structure and functionality.25
Next-generation sequencing and mass spectrometry have transformed the understanding of cancer biology by enabling comprehensive genomic, transcriptomic, and proteomic profiling. Next-generation sequencing allows for both genomic DNA and RNA sequencing.25 RNA sequencing explores transcripts, isoforms, splice variants, single-nucleotide polymorphisms, and chimeric gene fusions with high sensitivity and accuracy.17,22 Mass-spectrometry-based proteomics quantifies proteins in cells, tissues, and fluids.25 Large-scale initiatives such as The Cancer Genome Atlas (TCGA) programme integrate publicly available multi-omics databases across thousands of patients, providing a reference framework for NSCLC.21,26
Karaman et al.,15 conducted a network-based integrative analysis of RNA sequencing and DNA methylation data across lung, breast, colorectal, and kidney cancers identifying common prognostic biomarkers. This approach led to the identification of several significant biomarkers, including the SEC61G and the PTDSS1 genes associated with poor survival outcomes.15
Metabolomics has emerged as a powerful tool for identifying metabolic alterations.13 Shestakova et al.27 used targeted metabolomic profiling to distinguish patients with NSCLC from healthy individuals, identifying changes in tryptophan metabolism, the tricarboxylic acid cycle, and lipid metabolism, and developed a machine learning model with high diagnostic accuracy (area under the curve: 0.96), highlighting the potential of metabolomics in NSCLC diagnostics.27 Plasma and serum metabolomics offer minimally invasive diagnostics, but disease-specific plasma metabolites remain difficult to validate.13
Despite these advances, integration remains challenging. Multi-omics datasets are complex, heterogeneous, and high dimensional, requiring sophisticated computational methods.28-30
Machine learning and deep learning approaches are increasingly applied,31,32 but reproducibility and interpretability remain obstacles. Addressing these challenges demands advanced computational methods and substantial resources, but overcoming them could result in significant advancements in precision oncology and personalised medicine for NSCLC.33
This review quantitatively synthesises the predictive performance of integrated multi-omics models and evaluates the impact of open-source tools on translational research. By examining studies that leverage clinical data and multi-omics technologies to identify key biomarkers and relevant pathways in NSCLC, the author provides a roadmap for future clinical implementation, highlighting both critical challenges (specificity, reproducibility, context-dependence) and emerging opportunities in the field.
METHODS
For this systematic review, the author conducted a structured literature search using PubMed, covering publications from January 2020–March 2025. The objective was to identify original research articles reporting multi-omics data with clinically relevant biomarker findings in NSCLC. Three keywords-based strategies were applied using the ‘[Title] AND [Title/Abstract]’ filed tags to capture studies addressing omics modality, disease intersection, biomarker relevance, subtype stratification, and TME.
The search strings were:
- lung cancer and omics ([Title/Abstract]) AND (lung cancer [Title])) AND (omics [Title/Abstract];
- NSCLC and omics([Title/Abstract]) AND (NSCLC[Title])) AND (omics [Title/Abstract]; and
- NSCLC, omics, and resistance([Title/Abstract]) AND (omics [Title])) AND (resistance [Title/Abstract]).
Filters included “LUAD,” “LUSC,” and “tumour microenvironment” to refine results towards subtype-specific and clinically actionable findings. Additional keywords such as “biomarker,” “diagnostic,” “prognostic,” “therapeutic,” “survival,” and clinical outcome” were used to identify studies with translational potential.
Studies included were original research articles reporting multi-omics data on NSCLC biomarkers that focused on human subjects, provided subtype-specific insights (lung adenocarcinoma [LUAD], lung squamous cell carcinoma [LUSC]), and were published in peer-reviewed journals. The studies that were excluded were reviews, editorials, or conference abstracts, as well as studies lacking omics integration or clinical relevance.
The PubMed searches yielded 196, 30, and three records, corresponding to search one, two, and three, respectively. After deduplication and relevance screening, 30 articles were shortlisted, of which six were selected for detailed synthesis based on methodological rigour and translational value. To ensure currency and alignment with the review’s objectives, two additional studies published in March 2025 were manually incorporated into the final analysis (Figure 1).
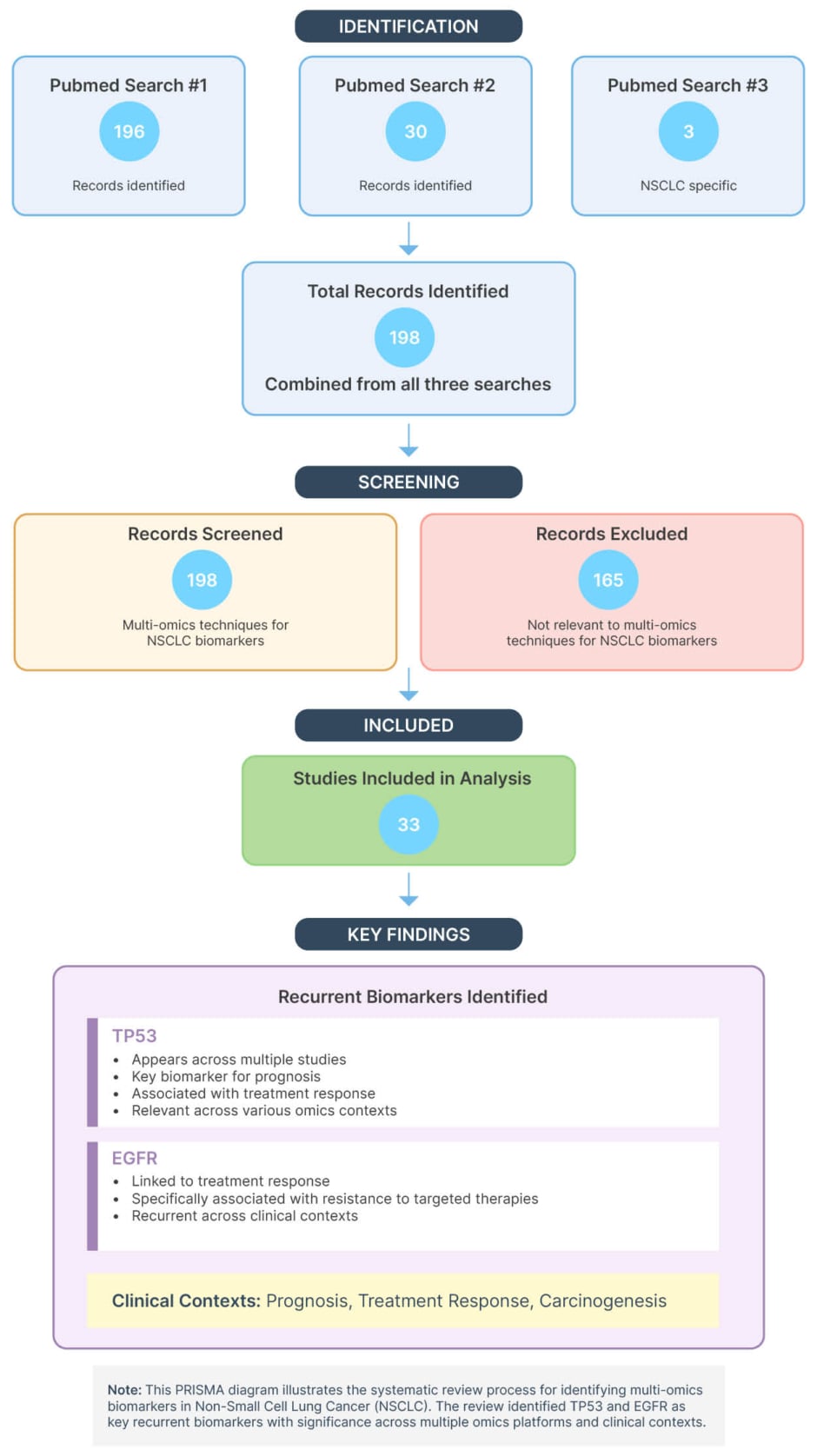
Figure 1: Preferred Reporting Items for Systematic reviews and Meta-Analyses flow diagram of study selection for non-small cell lung cancer multi-omics biomarker review.
NSCLC: non-small cell lung cancer; PRISMA: Preferred Reporting Items for Systematic reviews and Meta-Analyses.
All included studies were appraised for methodological clarity, completeness of omics layers, subtype stratification, and relevance to clinical endpoints. Bias was mitigated through predefined eligibility criteria, transparent screening, and standardised data extraction, with emphasis on studies demonstrating reproducibility, multi-layer integration, and external validation.
RESULTS
Multi-omics analyses identified a broad spectrum of biomarkers with diagnostic, prognostic, therapeutic, and mechanistic relevance in NSCLC. To enhance clarity and clinical utility, findings were synthesised into functional domains and molecular pathways, highlighting convergent drivers, resistance mechanisms, and survival determinants.
Oncogenic Drivers and Tumour Suppressors
Mutations in TP53 and EGFR were the most consistent signals across studies. TP53, the most frequently mutated gene in NSCLC,31 was associated with recurrence, immune evasion, and resistance to conventional therapies,22,31,33 positioning it as both a prognostic and predictive biomarker. Its mutation status also influenced sensitivity to immune checkpoint inhibitors.18,33,32 In LUAD compared with LUSC, TP53 mutations were linked to altered tumour microbiota, immune infiltration, and histological architecture, and could be predicted using multimodal deep learning models integrating histopathology, microbiome, and transcriptomic features.31
EGFR mutations, present in over half of early-stage NSCLC cases, predicted shorter disease-free survival and recurrence.32 EGFR-TKIs remain a cornerstone of precision medicine,34 and integrative multi-omics analyses confirmed favourable overall survival with afatinib in patients who were EGFR-positive.35 Resistance to EGFR-TKIs frequently emerged through secreted phosphoprotein 1 (SPP1) overexpression and activation of the yes-associated protein (YAP)/ transcriptional coactivator with PDZ-binding motif (TAZ) driven epithelial-mesenchymal transition (EMT), conferring stem-like and invasive properties.34,36,37 Other genomic signals included TTN mutations, correlating with chemotherapy and immunotherapy response,22,32 and ZNF71, which stratified patients into prognostic groups.38,39
Interpretation
TP53 and EGFR exemplify convergent drivers that shape both prognosis and therapy, while EMT-related pathways highlight recurrent mechanisms of therapeutic escape with the need for adaptive monitoring.
Immune Modulation and Systemic Inflammation
Immune-related biomarkers were prominent across omics layers, ZFHX3 mutations correlated with enhanced immune responses and immunotherapy sensitivity.36 Circulating cytokines such as IL6, IL8, CSF1, and C-X-C motif chemokine ligand 13 (CXCL13) predicted poor survival.8 Integrated microbiomic, metabolomic, and proteomic analyses identified C-reactive protein (CRP), lipopolysaccharide binding protein (LBP), and cluster of differentiation (CD)14 as systemic inflammatory markers reflecting systemic immune activation.40
Interpretation
Immune biomarkers provide stratification for immunotherapy, but also signal systemic inflammation, reinforcing the need for integrated host–tumour profiling.
Epigenetic and Metabolic Signatures
DNA methylation alterations (MGMT, CDKN2A, PCDH17, IRX1, TBX5, and HSPB6) were recurrently implicated in carcinogenesis.10,11 Large-scale, genomewide association studies linked methylation biomarkers to NSCLC risk,11 while epigenomic–transcriptomic integration nominated novel methylation biomarkers with diagnostic and therapeutic relevance.10
Metabolomic profiling differentiated LUAD from LUSC,41 while integrated metabolomic–proteomic analyses identified biomarkers predictive of immunotherapy sensitivity.33
Interpretation
Epigenetic and metabolic biomarkers highlight contextdependent signals that enrich stratification but require integration with genomic and transcriptomic data for reliable application.
Survival-Associated Biomarkers
Survival outcomes were linked to both tumour intrinsic signals and systemic mediators. Transcriptomic signals such as NKX21, CAV1, YBX1, FN1, and CDH3 were associated with LUAD survival.42 Circulating proteins (IL6, IL8, CSF1, matrix metallopeptidase 12 [MMP12], CXCL13) predicted poor outcomes.8 Immunerelated genes (TNS3, SEPT7, PUS1, IRF9, COMP, KLRB1, CD45, CD244) further correlated with survival.17,9,43,44
Interpretation
Survival reflects the dual importance of tumour biology and host immune response, reinforcing the need for integrated biomarker panels in prognostic modelling.
Longitudinal and Clinical Insights
Several studies emphasised the value of tracking biomarker patterns over time. Serial sampling and timeseries omics data enabled the monitoring of tumour evolution, the emergence of resistance, and the adaptive responses to therapy.31,34,45 Dynamic cytokine profiling revealed immune shifts,41 while comparative analyses distinguished NSCLC from sarcoma.42 Importantly, leptomeningeal metastasis was identified as a severe complication in EGFR-mutant NSCLC. Cerebrospinal fluid-based biomarkers enabled early detection/identified central nervous system (CNS) dissemination in EGFR-mutant LUAD and CNS-specific monitoring.8
Interpretation
Longitudinal profiling demonstrates how biomarkers evolve under therapeutic pressure, offering realtime insights into resistance and progression, and reinforcing the need for adaptive, multi-omics-guided clinical strategies.
Leptomeningeal metastasis highlights the importance of extending biomarker discovery to CNS involvement.
Integrated Insights
Synthesising across pathways reveals three dominant themes:
Convergent drivers (TP53, EGFR) underpin both prognosis and therapeutic response, making them central to biomarker-guided strategies.
Resistance mechanisms (SPP1, YAP/TAZ, EMT, inflammatory cytokines) recur across omics layers, highlighting the importance of longitudinal monitoring and adaptive therapy.
Context dependent signals (methylation, metabolic fingerprints, immune mediators) enrich stratification but require multilayer integration for reproducible application.
This pathway-based synthesis distils actionable conclusions from a data rich literature base, bridging molecular complexity with clinical translation. It highlights the need for biomarker frameworks that integrate tumour drivers, resistance pathways, and systemic host responses to guide precision oncology in NSCLC.
DISCUSSION
Recent years have witnessed a marked increase in studies applying multi-omics approaches to biomarker discovery in NSCLC. In the context of NSCLC, transcriptomics and genomics remain the dominant platforms, while proteomics and metabolomics provide complementary insights into treatment response and patient stratification.
Epigenomics, immunogenomics, and microbiomics play supporting roles in specific biomarker identification, enabling a more holistic molecular characterisation of NSCLC.
The advancements in biomarker discovery through multi-omics approaches hold great promise for clinical applications in NSCLC. In summary, the clinical outcomes associated with biomarker identification in the selected research studies can be applied to the following.
Prognosis
Multi-omics approaches have advanced prognostic assessment by extending beyond canonical oncogenes such as TP53 and EGFR to include immune-related factors (CXCL13, MMP12, and CSF-1) and metabolites as candidate biomarkers to predict disease progression and patients’ response to targeted treatments and immunotherapy.22,31-34 For example, TP53 mutations not only correlate with poor outcomes but also impair antigen presentation and modulate cytokine signalling, contributing to immune evasion and reduced immunotherapy efficacy.35 These findings highlight how integrated biomarker panels can refine risk stratification, although reproducibility across cohorts remains a critical challenge. Collectively, they reflect a broader shift towards understanding the tumour’s molecular complexity and its interactions with both the immune system and the TME, with the ultimate goal of advancing targeted therapy and patients’ stratification.35,46
Early-Stage Detection
Blood-based biomarkers offer a minimally invasive route for early lung cancer detection, but their translation is limited by specificity and reproducibility issues. Genomic signatures and network analyses have identified a 12-gene signature that can detect lung cancer in biological fluids at early stages of the disease and is associated with a poor disease outcome, enabling routine screening and early intervention to improve patient outcomes.38 Several studies also used multi-omics techniques (transcriptomics, genomics, and metabolomics) to identify biomarkers that differentiate NSCLC from normal tissue.39,47 Integrating metabolomics and lipidomics emphasises the importance of characterising the metabolome and lipidome in the plasma of patients with lung cancer to create a comprehensive metabolic fingerprint and identify potential clinical diagnostic markers for lung cancer.39,23
Therapy Response Prediction
Multi-omics platforms have identified biomarkers predicting therapy response, confirming the importance of oncogenes such as CCND1, TP53, MYC, and EGFR in identifying the susceptibility of NSCLC to therapies such as TKIs and immuno-checkpoint inhibitors.33,10 Furthermore, metabolomic and proteomic analyses refine immunotherapy response prediction,33 while SPP1 overexpression exemplifies how tumour-intrinsic changes remodel the TME to drive EGFR-TKI resistance, promoting tumour-associated macrophage infiltration and immunosuppressive signalling and ultimately leading to poor survival outcomes.40 Other candidates, such as ZFHX3 mutations, correlate with an increased immune response, suggesting predictive utility for immunotherapy in patients with lung cancer.11 Multi-omics platforms have identified predictors of therapy response, confirming that oncogenes (such as HMGB3 overexpression) are linked to worse survival in small cell lung cancer, while CASP10 is associated with better prognosis, indicating their potential as prognostic biomarkers. These examples underscore both the promise and the need for robust validation frameworks to ensure reproducibility.11
Chemotherapy andImmunotherapy Guidance
Combined metabolomic and transcriptomic analyses distinguish LUAD and LUSC subtypes, identify prognostic markers, and predict immunotherapy sensitivity.33,48 A proposed 14-gene signature serves as a biomarker panel to guide immunotherapy and chemotherapy, supporting personalised treatment for patients with NSCLC.45 Importantly, the integration of omics data into treatment algorithms must account for tumour heterogeneity and dynamic changes in therapy, reinforcing the need for longitudinal sampling.
Survival Prediction
Multi-omics approaches have revealed molecular markers associated with survival outcomes.37-41 Easily measurable serum proteins (e.g., IL-6, IL-8) offer non-invasive biomarkers for predicting poor outcomes,34 though their interpretive value is confounded by systemic inflammation and comorbidities. These findings highlight the potential of multi-omics to stratify patients by survival risk, while also underscoring the importance of context-dependent validation.
Carcinogenesis
Epigenomic studies highlight methylation-based biomarkers as potential diagnostic and therapeutic targets.8,42 Genome-wide association studies confirm that genetic and environmental factors shape methylation patterns, reinforcing their relevance in NSCLC development.8 However, methylation signatures remain highly variable and context dependent, influenced by tumour subtype, anatomical site, and immune status. This variability underscores the importance of multi-layered integration to capture the carcinogenic process more faithfully.
Translational Framework and Future Directions
The integration of multi-omics into clinically actionable endpoints remains challenging due to high dimensionality, heterogeneity, and computational complexity.28,30 Advanced machine learning and deep approaches31,32 are increasingly applied, but reproducibility and interpretability remain obstacles. The author’s proposed framework (Figure 2) maps five omics domains (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) onto diagnostic, prognostic, therapeutic, mechanistic, and longitudinal categories. By stratifying outputs across NSCLC subtypes, this framework supports biomarker discovery, resistance profiling, and personalised therapy guidance, reinforcing the trajectory from molecular insight to patient-centred outcomes.
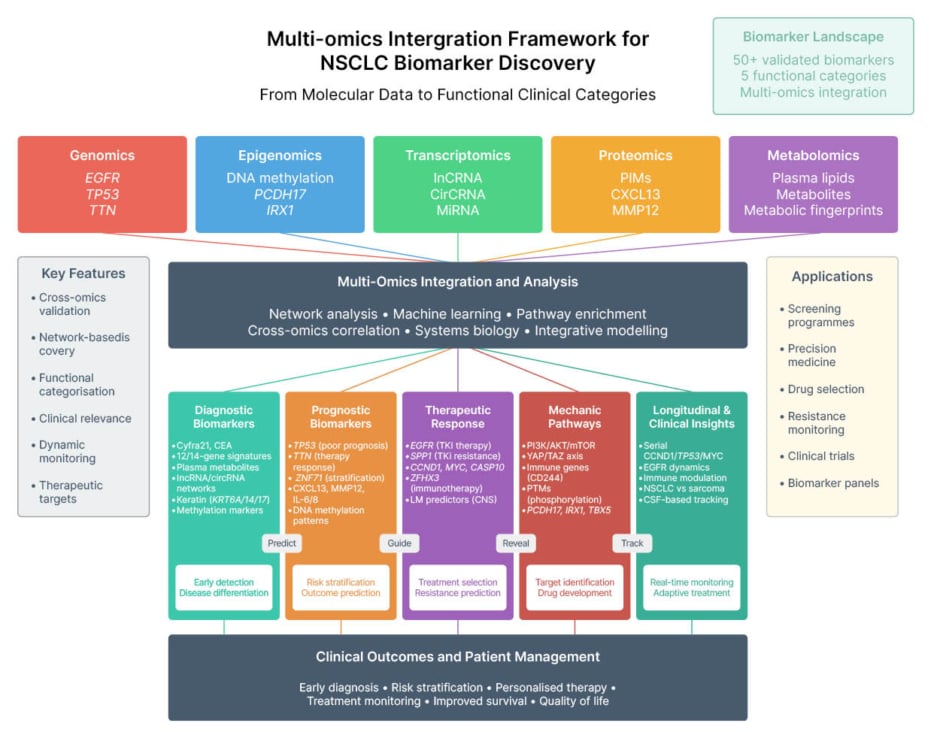
Figure 2: Multi-omics integration framework for non-small cell lung cancer biomarker discovery: mapping molecular inputs to functional clinical endpoints.
This layout reflects the progression from molecular input to patient-centred outcomes, supporting biomarker classification and clinical application.
AKT: protein kinase B; CEA: carcinoembryonic antigen; circRNA: circular RNA; CNS: central nervous system; CSF: cerebrospinal fluid; CXCL13: C-X-C motif chemokine ligand 13; Cyfra21: cytokeratin fragment 21-1; EGFR: epidermal growth factor receptor; IRX1: iroquois homeobox 1; LM: leptomeningeal metastasis; lncRNA: long non-coding RNA; miRNA: micro RNA; MMP12: matrix metallopeptidase 12; NSCLC: non-small cell lung cancer; PCDH17: protocadherin 17; PTM: posttranslational modifications; SPP1: secreted phosphoprotein 1; TAZ: transcriptional co-activator with PDZ-binding motif; TBX5: T-box transcription factor 5; TKI: tyrosine kinase inhibitor; vs: versus; YAP: yes-associated protein.
CONCLUSION
Multi-omics approaches are reshaping NSCLC research, transforming biomarker discovery, diagnosis, prognosis, and treatment optimisation. By integrating genomics, transcriptomics, proteomics, metabolomics, and epigenomics, these strategies move beyond single-layer analyses to capture the molecular complexity of tumours and their interactions with the microenvironment,22,31-34 enabling more precise patient stratification and accelerating the transition towards personalised therapeutic strategies.35-43
Recent advances highlight three major implications: multi-omics expands the repertoire of clinically actionable biomarkers,33,10 refines patient stratification to predict therapy responses,11,45 and decodes tumour complexity and resistance pathways to inform targeted interventions.40,41-47
Multi-omics approaches provide valuable insights into the molecular complexity of tumours and their interactions with the TME, but several challenges remain. The absence of standardised protocols, incomplete datasets, and the biological heterogeneity of NSCLC limit reproducibility and clinical translation.36,42 High-dimensional data demand advanced computational pipelines, rigorous statistical frameworks, and collaborative validation across diverse cohorts. Without these, the promise of multi-omics risks remaining confined to research settings.17,42
Incomplete Datasets
Missing data are common due to limitations in sample availability, costs, and experimental issues. These missing values can hinder data integration and compromise the validity of downstream analyses. To address this, researchers have developed sophisticated imputation methods that leverage the correlations among different omics layers to estimate missing data more accurately.9
Biological Complexity
Integrating these techniques remains challenging due to the heterogeneity among different cancer types and within tumours of the same type. An accurate interpretation of complex genetic and molecular data still faces barriers due to difficulties in integrating genomics, transcriptomics, proteomics, and metabolomics data from diverse sources.17,43 Handling multiple high-dimensional datasets and interpreting complex biological systems also represents a barrier, requiring advanced computational utilities and rigorous statistical methods to guarantee accurate data interpretation.17 Therefore, the identification of reliable biomarkers still poses a hurdle and requires the collaboration and combined efforts of different sources.17,43
Looking forward, open-source tools and collaborative research networks will be pivotal in bridging the gap between discovery and clinical implementation.9,17 By offering accessible, scalable, and reproducible analytical frameworks, open-source platforms empower researchers to integrate and analyse complex multi-omics datasets more effectively.
Advancing NSCLC care requires coordinated, multidisciplinary efforts to bridge the gap between research and clinical implementation, thereby accelerating progress in diagnostics, prognostics, and therapeutic strategies. The integration of multi-omics not only enriches understanding of tumour biology but also establishes a foundation for precision medicine. Sustained collaboration and innovation are essential to translate these insights into practice, ultimately improving patient outcomes and shaping the future of cancer care.
To operationalise multi-omics insights into patient-centric strategies, the author proposes a decision-making framework (Figure 3) that unifies five omics layers (genomics, transcriptomics, proteomics, metabolomics, and epigenomics) and maps them to actionable biomarkers, subtype-specific therapies, and resistance mechanisms. This model supports clinical decision-making by linking molecular profiles to targeted treatments, the prediction of immunotherapy sensitivity, and longitudinal monitoring, thereby reinforcing the translational trajectory from molecular discovery to personalised oncology.
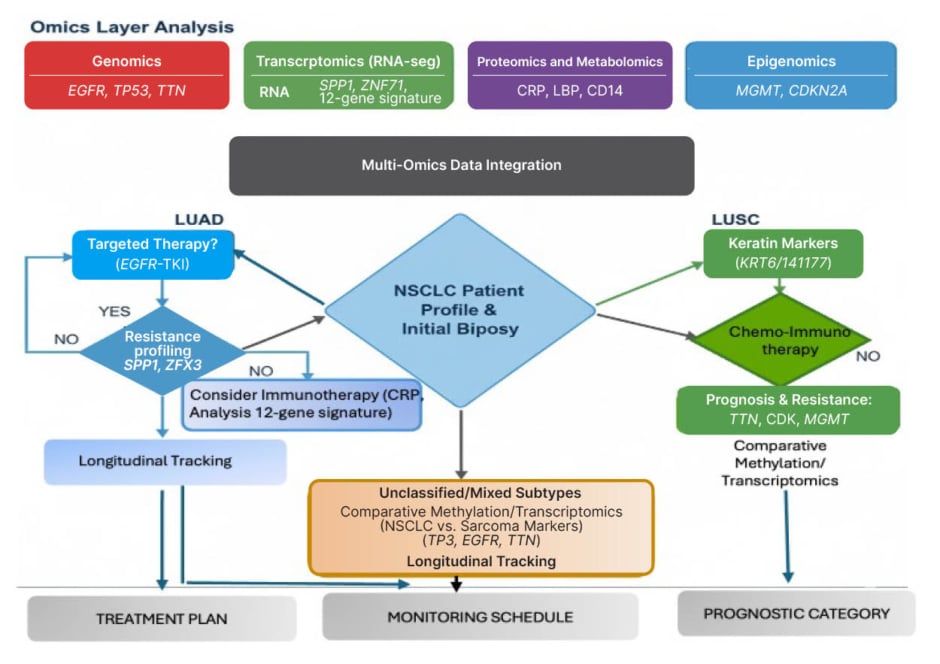
Figure 3: Multi-omics decision-making framework for non-small cell lung cancer: integrating molecular profiles to guide subtype-specific treatment and monitoring.
This patient-centric, decision-making framework for NSCLC integrates multi-omics data to inform subtype-specific therapeutic strategies. Molecular inputs, including genomics (EGFR, TP53, TTN), transcriptomics (RNA sequencing, gene expression, splicing), proteomics/metabolomics (protein expression, post-translational modifications, metabolites), and epigenomics (MGMT, CDKN2A), are consolidated through a centralised integration pipeline. The framework anchors on the initial biopsy and NSCLC patient profile, branching into tailored pathways for LUAD, LUSC, and unclassified/mixed subtypes. Each pathway incorporates molecular markers to guide treatment selection (e.g., EGFR-TKI, immunotherapy, chemo-immunotherapy), resistance profiling, and longitudinal tracking. Outcome categories include treatment planning, monitoring schedules, and prognostic stratification, reinforcing the translational trajectory from molecular insight to personalised care.
CD: cluster of differentiation; CRP: C-reactive protein; EGFR: epidermal growth factor receptor; LBP: lipopolysaccharide binding protein; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; NSCLC: non-small cell lung cancer; SPP1: secreted phosphoprotein 1; TKI: tyrosine kinase inhibitor; vs: versus; ZFX3: zinc finger protein x linked; ZNF71: zinc finger protein 71.