
Abstract
The immature oocytes within primordial follicles are arrested at Prophase I of meiosis and remain dormant until awakened by an increase in intracellular levels of phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Oocyte PIP3 level is determined by the balance between the activity of phosphoinositide 3-kinase (PI3K) and phosphatase and tensin homologue (PTEN). When this balance favours PI3K, PIP3 levels elevate and trigger the cascade of PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway, leading to activation of primordial follicles. This short review aims to provide new insights into the physiological functions of PI3K and PTEN in immature oocytes by summarising recent findings from murine model studies, including oocyte-specific transgenic mice with constitutively-active mutant PI3K.
OVARIAN RESERVE SIZE IS THE PREDOMINANT DETERMINANT OF FEMALE REPRODUCTIVE LIFESPAN
Female mammals are born with a finite number of oocytes, and the initial pool of primordial follicles (PF) is referred to as the ovarian reserve. The oocytes within the PF are arrested at Prophase I of meiosis and remain quiescent within the ovary until recruited into the growing follicle pool.1,2 Only a small number of PF are activated in each reproductive cycle, and this selective recruitment of PF is tightly regulated so that the reproductive lifespan is maintained for months in mice and decades in humans. If the activation rate of PF is accelerated, the ovarian reserve in women would be exhausted before reaching the age of expected menopause. The average age of women in the USA at the time of menopause is 51 years. If loss of normal ovarian function occurs before the age of 40 years, the condition is called premature ovarian failure or primary ovarian insufficiency (POI).3-5
BALANCE BETWEEN PHOSPHOINOSITIDE 3-KINASE AND PHOSPHATASE AND TENSIN HOMOLOGUE CONTROLS FOLLICLE ACTIVATION
The awakening of oocytes in PF is triggered by an increase in the levels of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) within the oocyte.6-9 The primary regulators of oocyte PIP3 levels are phosphoinositide 3-kinase (PI3K) and phosphatase and tensin homologue (PTEN), which exist in a counterbalanced relationship; PI3K catalyses phosphatidylinositol 4,5-bisphosphate (PIP2) to PIP3, whereas PTEN converts PIP3 back to PIP2 (Figure 1). When the balance favours PI3K activity, an elevated PIP3 level triggers the cascade of PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway: the binding of PIP3 to the pleckstrin homology domain anchors AKT to the plasma membrane; it is then phosphorylated and activated by phosphoinositide-dependent kinase 1 (PDK1). Phosphorylated AKT then targets multiple that control the activation of PF. Forkhead box O3 (FOXO3) transcription factor is one of the major direct substrates of AKT in immature oocytes. FOXO3 resides in the nucleus of dormant primordial oocytes, repressing the expression of genes that awaken the oocytes. When phosphorylated by phosphorylated AKT, FOXO3 is excluded from the nucleus and the expression or repression of FOXO3 target genes activates the oocyte.10,11 Phosphorylated FOXO3 remains in the cytoplasm of primary follicles until it is eventually degraded in secondary follicles.12 AKT also directly phosphorylates tuberous sclerosis (TSC) 2 and relieves the inhibitory effects of the TSC1-TSC2 complex on Ras homologue enriched in brain and mTOR complex 1 (mTORC1).
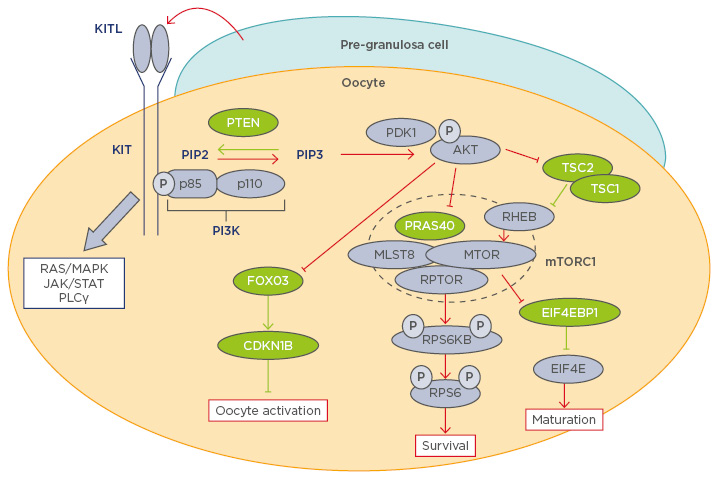
Figure 1: Diagram of the phosphoinositide 3-kinase pathway in oocyte physiology.
Molecules coloured green and purple are negative and positive regulators of the PI3K/AKT/mTORC1 pathway respectively. Red and green lines indicate activation and inhibition of the PI3K/AKT/mTORC1 pathway respectively.15-18,23-27,32-34,37
AKT: protein kinase B; CDKN1B: cyclin-dependent kinase inhibitor 1B; EIF4E: eukaryotic translation initiation factor 4E; EIF4EB1: eukaryotic translation initiation factor 4E binding protein 1; FOXO3: forkhead box 03; JAK: janus kinase; KITL1: KIT ligand 1; MAPK: mitogen-activated protein kinase; MLST8: mTOR associated protein, LST8 homologue; mTOR: mechanistic target of rapamycin; mTORC1: mechanistic target of rapamycin; p: phosphate group; PDK1: phosphoinositide-dependent kinase-1; PI3K: phosphoinositide 3-kinase; PLCγ: phospholipase C, gamma; PTEN: phosphatase and tensin homologue; PRAS40: proline-rich AKT1 substrate of 40 kDa; RHEB: Ras homologue enriched in brain; RPS6: ribosomal protein S6; RPS6KB: ribosomal protein S6 kinase B1; RPTOR: regulatory-associated protein of mTOR; STAT: signal transducer and activator of transcription; TSC: tuberous sclerosis.
The critical role of the PI3K/AKT/mTOR pathway in the regulation of PF activation was established by studies in murine genetic models in which the negative regulators of the PI3K/AKT/mTOR pathway are ablated in oocytes.9,13,14 In the ovaries of oocyte-specific Pten conditional knockout (cKO) mice,15 the ovarian reserve was completely depleted by postnatal day (PD) 28 through spontaneous activation of PF; however, the global premature activation of PF in Pten cKO mice was reversed by concomitant loss of PDK1.16 This study clearly indicates that the activity level of the PI3K pathway determines whether PF is activated or not. Similar to Pten cKO mice, oocyte-specific ablation of Tsc1 and Tsc2, the negative regulators of mTORC1, resulted in premature ovarian failure or POI. Both oocyte-specific Tsc1 cKO and oocyte-specific Tsc2 cKO mice replicated the phenotypes of oocyte-specific Pten cKO mice, and their ovarian reserve was depleted by PD28.17 Furthermore, Foxo3 null or oocyte-specific Foxo3 cKO mice also deplete the ovarian reserve through global premature follicle activation.11,15 In all null mutant mice for the negative regulators of the PI3K/ AKT/mTOR pathway, primary oocytes are activated almost immediately after the formation of PF, indicating that the cue for PF activation is a signal that disrupts the mechanism maintaining oocyte quiescence. Indeed, the oocyte expression of mutant FOXO3, which lacks critical phosphorylation sites for nuclear exclusion, slows down the recruitment of PF, suggesting that inactivation or phosphorylation of FOXO3 is not only sufficient but also essential to activate dormant oocytes.18 The critical role of PTEN activity in the maintenance of oocyte dormancy is not mouse specific. Dipotassium bisperoxo (5-hydroxypyridine-2-carboxyl) oxovanadate (bpV), a reversible inhibitor of PTEN, activates PF within human ovarian tissue fragments in vitro. The bpV treatment was accompanied with AKT phosphorylation and nuclear exclusion of FOXO3.19 Furthermore, the bpV treatment has led to successful deliveries of healthy babies in POI patients,14 establishing that mouse genetic models are valid models for human ovarian physiology.
MECHANISMS THAT CONTROL THE SELECTIVE RECRUITMENT OF PRIMORDIAL FOLLICLES
The exact mechanism that controls the selective activation of PF remains elusive. The generally accepted model proposes external signals that target pre-granulosa cells (GC) and/or oocytes trigger the signal transduction, leading to the activation of PI3K within oocytes.20-22 Using the analogy of an accelerator and brake may help explain the roles of PI3K (accelerator) and PTEN (brake) in PF activation (automobile): when the input of the accelerator exceeds that of the brake, the car moves forward (PF activation). When the brake is broken (PTEN null), the car can still crawl forward even without input from the accelerator, as revealed by spontaneous activation of PF in oocyte-specific Pten cKO mice. Furthering this analogy, mouse studies demonstrated the presence of multiple brakes in oocyte activation (e.g., TSC1, TSC2, FOXO3, and cyclin-dependent kinase inhibitor 1B) at different levels of the PI3K/AKT/mTOR pathway,16,18,22,23 and all brakes are simultaneously required to maintain the dormancy of primordial oocytes; PF are activated if any one of these negative regulators is lost.
In this regard, oocyte PTEN is the master brake maintaining the dormancy of PF because all other brakes in the system are released when PTEN activity is exceeded by the activity of PI3K. In theory, overproduction of PIP3 in oocytes should have the same effect as the loss of PTEN. Hence, we generated an oocyte-specific transgenic mouse model for a constitutively-active mutant PI3K (PIK3CA*, oo-Pik3ca* mice) and examined if the phenotypes mimic those of oocyte-specific null mice for PI3K/AKT/mTOR pathway inhibitors.24 Unexpectedly, overproduction of PIP3 by mutant PI3K was insufficient to activate the PF. Excess PI3K activity was counteracted by an up-regulation of PTEN. Correspondingly, the primordial oocytes retained FOXO3 in the nucleus and remained dormant. In accordance with these findings it was concluded that PTEN is dominant over PI3K activity in primordial oocytes and thus the activation of PF may involve a signal that down-regulates oocytic PTEN.
However, a recent genetic study in mice demonstrated the pivotal roles of the KIT–KIT ligand (KITL) signalling pathway in awakening dormant primordial oocytes, refuting our PTEN-dominant hypothesis. Oocyte-specific expression of the Kit Asp818Val allele, which produces a mutant KIT protein equivalent to the oncogenic mutant KIT Asp816Val in humans, spontaneously activated PF.25,26 Thus, the activation of the PI3K/AKT/mTOR pathway by KIT–KITL signalling is sufficient to awaken the oocytes within PF. The study strongly suggests that the recruitment of PF to the pool of growing follicles is triggered by the interaction between GC and oocytes, mediated by KIT–KITL signals. It raises the question that if activation of the PI3K signalling cascade is sufficient for the activation of PF, why does the expression of constitutively active PI3K not result in POI in mice? There are several nonexclusive explanations. One explanation for the contradictory phenotypes is the different level of PI3K activity and, thus, PIP3 concentrations (Figure 2). In conditional transgenic mice for KIT Asp816Val, the mutant Kit was knocked into the endogenous Kit locus, such that levels of mutant KIT Asp818Val protein and PI3K activation should be within physiological range. Therefore, the PIP3 levels should be elevated at a rate similar to innate follicle activation. In contrast, our transgenic mice overexpress the mutant PIK3CA* from the ROSA26 locus; as a result, the acute elevation of PIP3 to an unphysiological rate might have triggered an innate corrective system mediated by upregulation and nuclear accumulation of PTEN (Figure 2). A second explanation is the complexity of KIT signalling pathways. In addition to the PI3K/AKT/mTOR pathway, KIT activity is transduced by RAS/MEK/ERK, JAK/STAT, and phospholipase C-γ pathways.27 These signalling pathways may also play critical roles in the activation of primordial oocytes. Hence, the activation of PI3K alone does not replace KIT activity. Other pathways may indeed contribute to the repression of PTEN activity within oocytes.
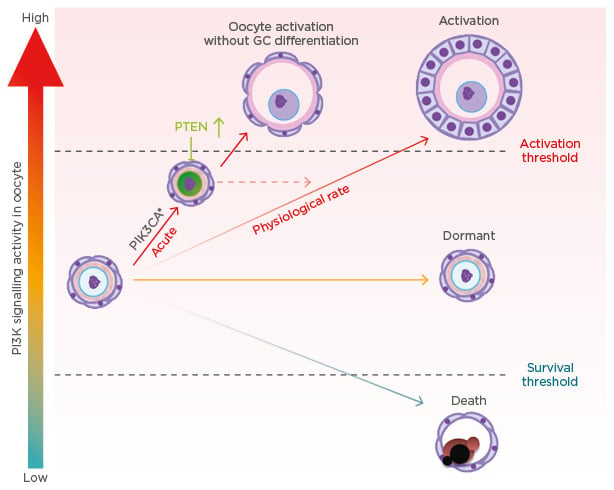
Figure 2: Oocyte phosphoinositide 3-kinase activity level and primordial follicle fate.
GC: granulosa cell; PI3K: phosphoinositide 3-kinase; PIK3CA*: constitutively active mutant PI3K; PTEN: phosphatase and tensin homologue.
INTRACELLULAR PHOSPHATIDYLINOSITOL (3,4,5)-TRISPHOSPHATE LEVELS ARE THE CRITICAL DETERMINANT OF OOCYTE SURVIVAL
The activation of Pik3ca* in primordial oocytes by Gdf9-iCre enlarged ovaries by retaining a higher number of follicles per ovary, including PF. This was unexpected, because the enlargement of ovaries in mutant mice of PI3K-mTOR negative regulators is due to synchronous activation of all PF, whereas the ovaries of oo-Pik3ca* mice contained the same number of PF as wild-type littermates at PD28. In female mice, apoptosis normally eliminates ˜80% of germ cells between meiotic entry and germ cell nest breakdown during embryogenesis.2 The apoptotic elimination of oocytes continues even after formation of PF during the first week of postnatal development. This naturally occurring selective-loss of oocytes is under the control of the TAp63α transcription factor encoded by Trp63; when Trp63 is conditionally ablated in oocytes by Gdf9-iCre, the number of PF per ovary at PD5 is doubled compared to normal ovaries.28 PF number in neonatal ovaries was also increased in null mutant mice of Bcl-2-associated X protein,29 phorbol-12-myristate-13-acetate-induced protein 1,30 p53 upregulated modulator of apoptosis,30,31 Bcl-2-like protein 11, and Bcl-2-modifying factor,30 the proapoptotic transcriptional targets of TAp63α in oocytes. The oo-Pik3ca* mice were defective in TAp63α-regulated selective-loss of oocytes in neonatal ovaries, and the apoptotic markers, such as cleaved poly (ADP-ribose) polymerase and Bcl-2-associated X protein, were significantly attenuated. The phenotypes of oo-Pik3ca* mice mimic those of mice defective in TAp63α-regulated oocyte apoptosis, and their follicle number at PD5 was twice that of normal ovaries.24 Hence, increase in PIP3 blocks the TAp63α-regulated oocyte apoptosis during neonatal development.
Although the inhibition of TAp63α-regulated apoptosis by PIP3 was unexpected, the PI3K/AKT/mTOR signalling pathway is known to play a key role in the survival of oocytes in follicles of all stages (Figure 1 and 2). For instance, oocyte-specific PDK1 null mice progressively lose ovarian follicles at any stage; however, the loss of oocytes in Pdk1 cKO mice was delayed by conditional deletion of Pten in the oocyte.33 Furthermore, an oocyte-specific null mutation in ribosomal protein S6 (RpS6), a downstream target of the PI3K/AKT/mTORC1/rpS6 kinase beta-1 signalling pathway (Figure 1), gradually lost ovarian follicles.33 Paradoxically, mTORC1 activity is dispensable for the normal folliculogenesis;34 the fertility remained normal in oocyte-specific deletion of Rptor gene encoding the regulatory-associated protein of mTOR (RAPTOR),35 which is essential for the assembly of mTORC1.36 Similarly, the ovaries of Pdk1 cKO mice and RpS6 cKO mice also contained growing follicles. The dispensability of key positive signalling molecules in the PI3K/AKT/mTOR pathway suggests the functional redundancies between the positive regulators. In fact, it has been suggested that loss of RAPTOR was compensated by increased PI3K.37 These observations contrast with the essentiality of negative regulator molecules of PI3K/AKT/mTOR pathway, suggesting the dominance of ‘brake’ over ‘accelerator’ in the activation of PF.
CONSEQUENCES OF EXCESS PHOSPHATIDYLINOSITOL (3,4,5)-TRISPHOSPHATE IN PRIMORDIAL OOCYTES
Even though the oocyte PI3K activity and PTEN levels were elevated, the activation of PF occurred normally in the oo-Pik3ca* mice, suggesting elevated PIP3 levels do not interfere with the innate mechanism directing PF activation (Figure 2). Meanwhile, the ovaries of oo-Pik3ca* mice contained abnormal follicles consisting of an enlarged oocyte and pre-GC. In normal folliculogenesis, the activation of oocytes and transformation of squamous pre-GC to cuboidal GC is synchronised; however, the enlarged oocytes in the abnormal follicles were activated, as they lacked expression of FOXO3 and TAp63α in the nucleus but expressed the zona pellucida glycoprotein 1 and 3.24 Since follicles are classified primarily by the morphology of GC, the abnormal follicle was designated as PF with activated oocytes. The absence of PF with activated oocytes in normal ovaries suggests the transformation of pre-GC to GC is a prerequisite for the activation of oocytes. Activation of KIT signalling pathways in oocytes by GC through KITL is likely involved in co-ordinating the activation of oocytes and pre-GC during normal folliculogenesis.25 Conversely, the asynchronous activation of oocytes and pre-GC in oo-Pik3ca* mice indicates that primordial oocytes can be activated independent of GC signalling when PI3K is constitutively active in oocytes. It has been speculated that the elevated PIP3 level allows oocytes to cross the activation threshold without stimuli from GC (Figure 2).
In the oo-Pik3ca* mice, antral follicles with developmentally competent oocytes accumulated in the ovaries. The cumulus-oocyte complexes collected from oo-Pik3ca* mice were capable of resuming meiosis when subjected to an in vitro maturation test. Despite the competence of follicles, oo-Pik3ca* mice were anovulatory and sterile. Since ovulation occurred normally with superovulation treatment, the anovulation was not due to defective oocytes or follicles, but rather a defective endocrine system caused by an excessive number of overgrown follicles. In oo-Pik3ca* mice, the level of follicle stimulating hormone was significantly low, while the serum concentration of both inhibin A and B were significantly increased. The altered hormones levels suggested defects in endocrine control were due to overgrown follicles.24
Interestingly, oo-Pik3ca* mice had significantly larger antral follicles due to an increased proliferation rate in GC, suggesting a deregulation of growth control in GC by the expression of PIK3CA* in the oocytes. The excessive number of overgrown follicles with elevated oocyte PIP3 had an unexpected consequence in 80-day-old mice: the development of GC tumours (GCT) of the ovary. The oo-Pik3ca* mice became cachectic by the age of 80 days due to bilateral GCT secreting activin A. The study38 suggested that local interactions with PIK3CA*-positive oocytes during early folliculogenesis establish an activin A-mediated autocrine growth circuit programme in GCT. This explains the mechanism of GC hyperplasia in antral follicles. Meanwhile, the molecular mechanisms underlying the transformation of GC to GCT in the oo-Pik3ca* mice are yet to be elucidated.
CONCLUSIONS
The aforementioned oo-Pik3ca* murine model has helped elucidate the physiological roles of the balance between PI3K and PTEN within oocytes. For instance, the expression of PIK3CA* prevented the activation of TAp63α and the downstream proapoptotic signalling pathway, resulting in a higher number of surviving PF. However, it is unclear how TAp63α activity in the selective loss of PF is regulated and how PI3K activity interferes with the activation of TAp63α. In regard to the mechanism of oocyte activation, the constitutively active KIT mutant model established the sufficiency of KIT–KITL pathways to activate PF. If KIT signalling triggers the PF activation in normal folliculogenesis, the KIT–KITL pathways should be activated only in a certain number of PF in each reproductive cycle; how KIT signalling activity is controlled in the selective recruitment of PF is an intriguing question. It is also unclear if the activation of PI3K is sufficient to transduce KIT signalling in primordial oocytes. Further research is required to answer these questions.






