Abstract
The multitude of research clarifying critical factors in embryonic organ development has been instrumental in human stem cell research. Mammalian organogenesis serves as the archetype for directed differentiation protocols, subdividing the process into a series of distinct intermediate stages that can be chemically induced and monitored for the expression of stage-specific markers. Significant advances over the past few years include established directed differentiation protocols of human embryonic stem cells and human induced pluripotent stem cells (hiPSC) into human kidney organoids in vitro. Human kidney tissue in vitro simulates the in vivo response when subjected to nephrotoxins, providing a novel screening platform during drug discovery to facilitate identification of lead candidates, reduce developmental expenditures, and reduce future rates of drug-induced acute kidney injury. Patient-derived hiPSC, which bear naturally occurring DNA mutations, may allow for modelling of human genetic diseases to enable determination of pathological mechanisms and screening for novel therapeutics. In addition, recent advances in genome editing with clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 enable the generation of specific mutations to study genetic disease, with non-mutated lines serving as an ideal isogenic control. The growing population of patients with end-stage kidney disease is a worldwide healthcare problem, with high morbidity and mortality rates, that warrants the discovery of novel forms of renal replacement therapy. Coupling the outlined advances in hiPSC research with innovative bioengineering techniques, such as decellularised kidney and three-dimensional printed scaffolds, may contribute to the development of bioengineered transplantable human kidney tissue as a means of renal replacement therapy.
INTRODUCTION
Decades of developmental studies have elucidated the molecular pathways and genes necessary for normal kidney development. Historically, these studies enabled an understanding of human kidney developmental disease processes but largely have not translated into effective treatment, as human kidney development ceases prior to birth. There has been a renaissance in developmental biology with the advent of research involving human pluripotent stem cells (hPSC). By definition, hPSC have the ability to differentiate into cells of the three germ layers, namely the mesoderm, ectoderm, and endoderm.1,2 Through directed differentiation, the sequential application of growth factors at specific concentrations for defined periods of time, hPSC can be transformed into particular organ tissues with a high degree of efficiency. Human induced pluripotent stem cells (hiPSC), a subset of hPSC, are generated from reprogramming adult cells through the activation of transcription factors characteristic of pluripotency.2,3 Starting with any individual’s terminally differentiated cells, an unlimited supply of isogenic hiPSC can be generated. Genomic retention permits modelling of genetic disease and provides an immunocompatible cell source for organ regeneration. The known physiology of vertebrate organogenesis serves as a guide towards the directed differentiation of hPSC into human tissue. Drawing from studies of vertebrate kidney development, researchers have discovered directed differentiation protocols that derive human kidney tissue in vitro.4-10 Such human kidney tissue in a dish, potentially coupled with advances in biomedical engineering, may prove to revolutionise the fields of drug discovery, disease modelling, and kidney regenerative medicine (Figure 1).11
Figure 1: Translational applications of hiPSC-derived kidney organoids.
The discovery of directed differentiation protocols for the generation of three-dimensional kidney organoids from hiPSC may provide for numerous translational applications, including human genetic and congenital disease modelling, kidney regenerative medicine, and nephrotoxicity screening during drug development.
hiPSC: human induced pluripotent stem cells.
VERTEBRATE KIDNEY DEVELOPMENT
The vertebrate kidney derives from the intermediate mesoderm (IM) of the mesodermal germ layer. During kidney organogenesis, the IM sequentially gives rise to the pronephros, mesonephros, and metanephros. In humans, the pronephros remains non-functional, regressing by the fourth week of gestation. The mesonephros forms just prior to degeneration of the pronephros in humans and serves as the primary excretory organ from the fourth to the eighth week of gestation. In females, the mesonephros degenerates, whereas in males it gives rise to portions of the gonads. The metanephros, which begins to form caudal to the mesonephros in the fifth week of gestation, becomes the definitive adult kidney in humans (Figure 2A).12
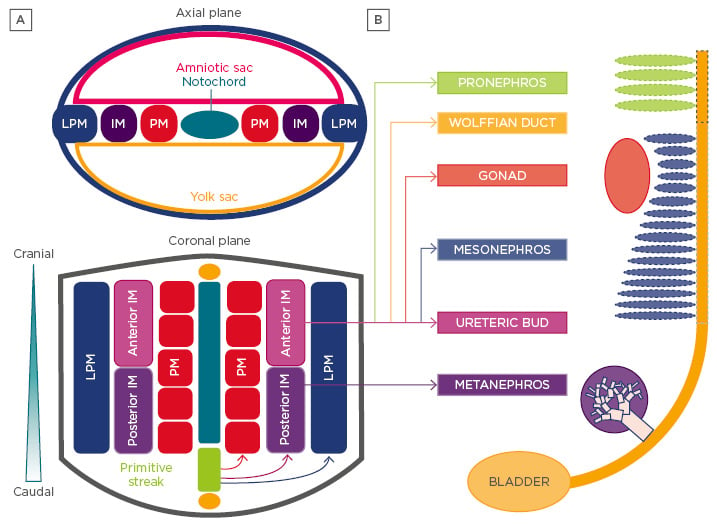
Figure 2: Development of the vertebrate kidney.
A) Vertebrate kidney development involves the serial progression of three distinct structures derived from the intermediate mesoderm, the pronephros, mesonephros, and metanephros. The pronephros is non-functional and regresses, the mesonephros provides primary exocrine functions until the eighth week of gestation, and the metanephros becomes the mature kidney in humans. Notably, the caudal portion of the mesonephric duct forms portions of the gonads in the male, while regressing in females. Each nephric stage associates with a collecting duct network derived from the Wolffian (or nephric) duct. B) The ureteric bud is an outpouching of the Wolffian duct, which derives from the anterior intermediate mesoderm. The metanephric mesenchyme contains the progenitor cells of all kidney epithelia, save the collecting duct, and derives from the posterior intermediate mesoderm.
LPM: lateral plate mesoderm; IM: intermediate mesoderm; PM: paraxial mesoderm.
The metanephric kidney forms through the reciprocally inductive interactions between two distinct IM tissues, the metanephric mesenchyme (MM) and ureteric bud (UB).4 The MM arises from the posterior IM and contains a population of multipotent nephron progenitor cells (NPC) that express the transcription factors Six2, Cited1, Pax2, Sall1, and Wt1.4,13-15 The Six2+ NPC cluster to form the cap mesenchyme around each infiltrating UB tip.15 UB-derived Wnt signals induce NPC to undergo mesenchymal-to-epithelial transition, giving rise to nearly all the epithelial cells of the nephron except for those of the collecting duct.15,16 Meanwhile, the UB arises as an epithelial outpouching from the caudal Wolffian (or nephric) duct, which upon receiving inductive signals from the MM, undergoes iterative branching to form the collecting duct system of the kidney. Nephrogenesis in humans is completed between 32 and 36 weeks of gestation and results in the formation of approximately one million nephrons in each kidney. After birth, no new nephrons are formed, even under circumstances of kidney injury and repair.17,18
Recent work from Taguchi et al.4 has provided important insight into the embryonic origins of NPC in the MM. Employing lineage tracing techniques in mice, the authors demonstrated that NPCs are derived from a population of T+ cells in the primitive streak that persists to give rise to T+Tbx6+ posterior nascent mesoderm followed by Wt1+Osr1+ posterior IM. In contrast, the UB originates from a Pax2+ cell population known to be limited to the anterior IM14,19,20 and is incapable of giving rise to MM (Figure 2B). Thus, careful consideration of these diverging developmental pathways is critical for the efficient differentiation of pluripotent stem cells (PSC) into cells of these two different lineages.
Current strategies to direct the differentiation of hPSC into cells of the kidney lineage have been based on vertebrate animal kidney development models.4,7,8 Key transcription factors, growth factors, and membrane protein factors involved in kidney organogenesis have been identified using gene knockout and transgenic models with resultant phenotype demonstrating congenital anomalies of the kidney and urinary tract (Table 1).21-38 These factors serve as critical markers of kidney induction during directed differentiation of hPSC.
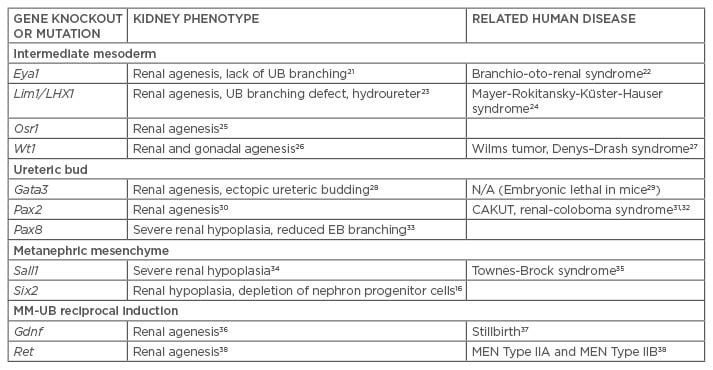
Table 1: Genetic knockout and transgenic mouse models of kidney development.
UB: ureteric bud; MM: metanephric mesenchyme; EB: embryoid bodies; CAKUT: congenital anomalies of the kidney and the urinary tract; MEN: multiple endocrine neoplasia.
PLURIPOTENT STEM CELLS
PSC represent early embryonic progenitor cells, believed to correspond to the blastocyst or epiblast stage of mammalian embryos.39 Cells at this stage arise 5–9 days post-conception in humans and are defined by two intrinsic properties: self-renewal and pluripotency. PSC have the ability to self-renew indefinitely in culture, without transformation or differentiation. Additionally, PSC are pluripotent, having the capacity to give rise to all cell types derived from the three embryonic germ layers, namely the mesoderm, endoderm, and ectoderm.40
PSC comprise embryonic stem cells (ESC) and induced pluripotent stem cells (iPSC). ESC are derived from the isolation and culture of cells from the inner cell mass of the embryonic blastocyst.1 In contrast, iPSC are generated following the activation of four key transcription factors of pluripotency (Oct4, Sox2, Klf4, and c-Myc) in terminally differentiated cells, which directly reprograms them into cells that behave similarly to and appear morphologically identical to ESC.2,3
Although human ESC (hESC) remain the gold standard for stem cell research, hiPSC have a number of advantages. Unlike hESC, the derivation of iPSC does not involve the use of human embryos, a limitation that has led to ethical concerns over the use of hESC.41 Protocols now exist to derive hiPSC from a variety of different terminally differentiated cell types, including peripheral blood mononuclear cells, keratinocytes, hepatocytes, urothelial cells, neural stem cells, and kidney mesangial and tubular epithelial cells, using both viral and non-viral reprogramming methods.42-48 hiPSC can be generated from any human being, healthy or diseased, with retention of the host’s individual genome. Therefore, hiPSC represent an isogenic substrate to generate theoretically immunocompatible tissue for organ regeneration. Additionally, hiPSC generated from patients with genetic diseases can be used to develop in vitro models to better study disease pathogenesis. For diseases that are particularly rare or do not have relevant animal models, iPSC offer a novel strategy to study disease mechanisms and develop new therapeutics.
In the absence of exogenous growth factors or chemicals, PSC undergo stochastic differentiation into embryoid bodies (EB) in vitro and teratomas in vivo.1-3 Both EB and teratomas are heterogeneous tissues that contain cells of the three embryonic germ layers. The heterogeneity of the tissue confirms pluripotency, but imparts a low induction efficiency of any one specific cell type. Directed differentiation refers to the process by which PSC are sequentially treated with growth factors and chemicals to efficiently induce a particular cell or tissue type of interest. Directed differentiation protocols often employ a stepwise approach, through intermediate developmental stages, which mirror normal embryonic organogenesis.49
Differentiation of Human Pluripotent Stem Cells to Kidney Lineage Cells
Chronologically, the earliest work to obtain IM from hESC involved the generation of WT1+ and PAX2+ kidney precursor cells.50,51 Shortly thereafter, protocols to directly generate cells bearing markers of terminal kidney epithelia were published. Podocin+Synaptopodin+PAX2+ podocyte-like cells were induced in EB from hiPSC using a combination of activin, bone morphogenetic protein (BMP)7, and retinoic acid.52 Through a similar protocol, a monolayer culture of these podocyte-like cells was shown to integrate into WT1+ glomerular structures, when combined with dissociated-reaggregated embryonic mouse kidneys. In renal epithelial growth medium, a combination of BMP2 and BMP7 induced the differentiation of hESC to aquaporin-1 (AQP1)+ proximal tubule-like cells. Flow-sorted AQP1+ cells integrated into tubular compartments of ex vivo newborn mouse kidneys and spontaneously formed cord-like structures when cultured on Matrigel®. AQP1+ cells increased cyclic adenosine monophosphate production in response to exogenous parathyroid hormone, demonstrated functional γ-glutamyl transferase enzymatic activity, and produced ammonia.53
Recent studies have focussed on the efficient induction of kidney progenitor cells, particularly cells of the IM and MM. An OSR1-GFP hiPSC line was treated with the combination of the glycogen synthase kinase-3β inhibitor CHIR99021 (CHIR) and activin, followed by BMP7, to yield OSR1+ cells with 90% efficiency within 11–18 days of differentiation. OSR1+ cells differentiated into populations expressing markers of mature kidneys, adrenal glands, and gonads in vitro and integrated into dissociated-reaggregated embryonic mouse kidneys.54 The sequential treatment of hPSC with CHIR, followed by fibroblast growth factor (FGF)2 and retinoic acid, generated PAX2+LHX1+ IM-like cells with >70% efficiency. PAX2+LHX1+ cells stochastically differentiated to form ciliated tubular structures expressing the proximal tubular markers Lotus tetragonolobus lectin (LTL), N-cadherin, and kidney-specific protein.6 Meanwhile, directed differentiation of PAX2+LHX1+ cells, involving treatment with FGF9 and activin, generated cells co-expressing markers of MM including SIX2, SALL1, and WT1.6 A similar protocol efficiently generated PAX2+LHX1+ IM cells from hESC within 6 days, using a combination of CHIR and FGF9.5 On continued FGF9 treatment, these cells gave rise to SIX2+ cells with 10–20% efficiency within 14 days.5 Mixing these cells with dissociated-reaggregated mouse embryonic kidneys resulted in three-dimensional (3D) aggregates of SIX2+ cells containing tubular structures expressing kidney markers such as AQP1, AQP2, JAG1, E-cadherin, WT1, and PAX2.5
While considerable work has been done to differentiate hPSC into MM, efforts to differentiate hPSC into cells of the UB lineage have been limited. In a 4-day directed differentiation protocol involving initial treatment with BMP4 and FGF2, followed by retinoic acid, activin, and BMP2, hESC, and hiPSC formed PAX2+OSR1+WT1+LHX1+ IM-like cells.55 These cells spontaneously upregulated transcripts of the UB markers HOXB7, RET, and GFRA1 within 2 days. Upon co-culture with dissociated-reaggregated embryonic mouse kidneys, these putative UB progenitor-like cells partially integrated into mouse UB tips and trunks.55
Two groups have demonstrated the ability to differentiate hPSC into 3D kidney organoids containing complex, multi-segmented nephron-like structures.7,8 Treatment of hPSC with CHIR for 4 days, followed by FGF9 for 3 days, and transfer into 3D suspension culture ≤20 days generated kidney organoids consisting of nephron-like structures bearing markers of proximal and distal tubules, early loops of Henle, and podocyte-like cells.7 To simulate UB-derived Wnt signalling, a transient 1-hour pulse of CHIR on transfer to suspension culture aided the induction of nephron-like structures. The organoids contained tubular structures expressing markers of collecting ducts, stromal cells expressing markers of the renal interstitium, and endothelial cells, suggesting the presence of a heterogenous mixture of the IM and lateral plate mesoderm.7 More recently, a directed differentiation protocol was found to differentiate both hESC and hiPSC into SIX2+SALL1+PAX2+WT1+ NPC that could be induced to form nephron (kidney) organoids in both two-dimensional and 3D cultures.8 In a stepwise approach that mirrored vertebrate kidney organogenesis, first T+TBX6+ primitive streak cells were induced with CHIR for 4 days, then WT1+HOXD11+ posterior IM cells were induced with activin, and then SIX2+SALL1+PAX2+WT1+ NPC were induced using low-dose FGF9 with ≤90% efficiency. Treatment of NPC with continued FGF9 and a transient CHIR pulse induced PAX8+LHX1+ renal vesicles that spontaneously formed nephron-like structures in two-dimensional culture. Transfer of NPC into 3D suspension culture resulted in the formation of kidney organoids containing multi-segmented nephron-like structures expressing markers of glomerular podocytes (NPHS1+PODXL+WT1+), proximal tubules (LTL+CDH2+AQP1+), loops of Henle (CDH1+UMOD+), and distal tubules (CHD1 UMOD–) in a contiguous arrangement. Additionally, these kidney organoids demonstrated promise for applications involving studies of kidney development and drug toxicity.
The establishment of efficient protocols for directing the differentiation of hPSC into NPC and kidney organoids marks a significant advance in the ongoing effort to apply human stem cells to the regeneration of kidney tissue, modelling of human kidney disease, and drug testing for therapeutic efficacy and toxicity.8,11 However, the development of definitive functional assays and the establishment of reliable genetic markers will be required to verify whether induced hPSC-derived kidney cells and tissues are sufficiently identical to their in vivo complements for translational applications.
KIDNEY ORGANOIDS FOR NEPHROTOXICITY TESTING
Nephrotoxicity is a common manifestation of the toxic effects of drugs and their metabolites. The kidneys are highly vascularised, receiving 20–25% of the cardiac output, and may accumulate circulating toxins in the vascular, interstitial, tubular, or glomerular spaces. During drug development, 19% of failures in Phase III clinical trials are due to nephrotoxicity.56 As the cost to bring a drug to market is currently ~2.6 billion dollars,57 the availability of high-throughput nephrotoxicity screening systems during drug development may save considerable time and costs.
Recent reports have demonstrated that hPSC-derived kidney cell tissue may respond to nephrotoxic drugs in a manner that mimics in vivo kidney injury.7,8 In the previously discussed protocols to generate hPSC-derived kidney organoids, the chemotherapeutic agent cisplatin was demonstrated to cause specific injury to the proximal tubular cells, consistent with known cisplatin toxicity in vivo. Cisplatin-induced proximal tubular injury was characterised by the upregulation of the DNA damage marker γH2AX, increased expression of kidney injury molecule-1 (KIM-1), and upregulation of cleaved caspase-3.7,8,10 Additionally, treatment of kidney organoids with the antibiotic gentamicin upregulated KIM-1 in proximal tubules, without any discernible effect on podocytes, consistent with the known nephrotoxicity of aminoglycosides.
KIDNEY ORGANOIDS FOR MODELLING OF KIDNEY DISEASES
hiPSC-derived organ tissue represents a valuable platform for the study of human pathophysiology and the discovery of novel therapeutics. As hiPSC remain isogenic with the original host cell prior to reprogramming, they provide a means of modelling patient-specific genetic diseases. Importantly, the rarity of many genetic diseases precludes enrolment in clinical trials, which coupled with a lack of incentive for drug companies to develop treatments for rare diseases, fuels the hope that hiPSC-based assays can be a scalable and reliable option for preclinical studies at low cost. Once established, reliable human disease models may allow for clinical ‘trials-in-a-dish’. Human stem cell-based systems may ultimately replace animal testing, known to be poorly predictive of the human response.58 To date, hiPSC lines have been generated for autosomal dominant polycystic kidney disease (ADPKD),59 autosomal recessive polycystic kidney disease (ARPKD),43 and systemic lupus erythematosus.60,61
ADPKD is the most common potentially lethal monogenic disorder, affecting 1 in 600 to 1 in 1,000 live births. Approximately 50% of individuals with ADPKD develop end-stage kidney disease (ESKD) by 60 years of age.62 The traditionally used mouse models are homozygous carriers for ADPKD mutations while afflicted humans are heterozygotes, calling into question the utility of ADPKD animal models.63 hiPSC lines have been created from multiple patients with ADPKD and ARPKD.59 A subsequent study from the same group used the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system to knockout or PKD2, the causative genes for ADPKD, in hESC lines.64 Tubular organoids derived from these PKD1 and PKD2 knockout mutants developed cystic structures from LTL+ kidney tubules, suggesting that this model could potentially serve as a novel means to study cystogenesis in ADPKD and screen for new therapeutics in vitro.
PLURIPOTENT STEM CELLS FOR BIOENGINEERING KIDNEY TISSUE
The shortage of transplantable organs, coupled with the rising prevalence of ESKD, have led researchers to apply regenerative medicine techniques to kidney bioengineering.65 Human iPSC serve as a theoretically immunocompatible and scalable cell source, with therapeutic applications for both chronic kidney disease and acute kidney injury. While the kidney is a complex organ consisting of >50 distinct cell types that provide for exocrine, endocrine, and metabolic functions, the essential elements of an envisioned bioengineered kidney would include multiple hPSC-derived cell types supported in a perfusable scaffold that provides appropriate cellular segregation and compartmentalisation. Two scaffolding approaches have been undertaken, kidney decellularisation and a 3D-printed framework.66,67
Decellularised kidney approaches preserve the extracellular matrix (ECM) of distinct kidney compartments, retaining matrix-associated signals and growth factors, and conserving the vascular tree and branched collecting duct network. Mammalian kidneys have been decellularised, using the detergent sodium dodecyl sulfate and the cell membrane toxicant Triton X-100, with haematoxylin and eosin stains confirming the removal of cellular material and immunohistochemistry demonstrating the preservation of native ECM.68 Surgically implanted decellularised vertebrate kidneys remain perfusable and lack blood extravasation, but completely thrombose due to denuded ECM.69 Seeding decellularised rat kidneys with rat fetal kidney cells via the ureter, and endothelial cells via the renal artery, enabled the production of a small amount of urine when perfused by the recipient’s circulation following orthotopic transplantation.66 However, the urinary filtrate was negligible and vasculature rapidly thrombosed. Similarly, murine ESCs were seeded into decellularised rat kidneys via the renal artery and ureter.70 Cells lost their pluripotent phenotype, expressed kidney markers, but were limited by small vessel thrombosis on perfusion testing in vivo. Decellularised vasculature ex vivo lined with the biocompatible polymer, poly(1,8-octanediol citrate), and functionalised with heparin, reduced platelet adhesion and whole blood clotting.71 Given their advantage of maintained architecture, decellularised kidney approaches may provide a valuable resource in efforts to create a bioengineered kidney.
Biologic applications of 3D printing have gained notoriety and credibility with the ‘organ-on-a-chip’ series, modelling lung, gut, the kidney proximal tubule, and bone marrow.72-75 While these early organ chips employ a soft lithography method first published nearly two decades ago,76 recent advances in 3D printing have enabled faithful manufacturing of micrometre scale, multicomponent 3D structures. However, current commercially available 3D printing resins, for both stereolithography and multi-jet modelling, demonstrate poor biocompatibility.77,78 To overcome obstacles of resin cytotoxicity and the need for a vascular network in tissue engineering, the Lewis lab employed biocompatible collagen-based resins to develop vascular networks.79 Human umbilical vein endothelial cells, embedded in a sacrificial Pluronic® F127 hydrogel, were printed in channels and surrounded by photocurable gelatin methacrylate. Removal of the fugitive ink yielded tubular channels consisting of a confluent monolayer of human umbilical vein endothelial cells. Using a similar methodology, the proximal tubule-on-a-chip has evolved from incorporating a cellular monolayer into tubular structures containing a perfusable lumen.67
DISCUSSION
Acclaim for human stem cell-based research has been based on its potential to influence patient care through translational applications. ‘Hope-based’ notoriety has preceded evidence-based credibility. Clinical trials using hiPSC-derived tissues are currently underway in patients with retinal disease and acute spinal cord injury. Concerning kidneys, recent protocols to generate 3D, multicellular, and multi-compartmentalised kidney tissue from hPSC has reinvigorated the promise of human stem cell work to revolutionise the fields of drug discovery, disease modelling, and regenerative medicine.
Nephrotoxicity is a common cause for failures during drug development, accounting for 19% of Phase III clinical trial failures.56 The proximal tubule is the predominant site of toxicity, where endogenous and exogenous toxin excretion is mediated by organic anion transporters (OAT) and organic cation transporters (OCT). Current methods to assess nephrotoxicity, involving immortalised human cell lines and animal models, are limited by inadequate expression and interspecies heterogeneity among OAT/OCT transporters, respectively. Use of hPSC-derived kidney tissue in vitro may prove to be an improved model. Future work to determine the OAT/OCT expression profiles of hPSC-derived kidney tissue, generated from the various directed differentiation protocols, may identify an optimal method to predict nephrotoxicity in preclinical studies, thereby preventing late phase failures. Confirmatory functional assays may include the reproduction of cisplatin proximal tubule toxicity, mediated by OCT2, with reversal using cimetidine, an OCT2 inhibitor. Reporter lines for KIM-1, a proximal tubule injury marker, and neutrophil gelatinase-associated lipocalin, a distal nephron injury marker, may permit real-time toxicity monitoring. Adapting human kidney tissue in vitro to perfusable systems, simulating in vivo conditions, may improve the sensitivity and specificity of identifying nephrotoxins through increased drug transporter expression. Additionally, perfusable systems would permit toxicity testing of individual compounds across increasing concentration gradients, greatly consolidating the test conditions required by preclinical testing methods.
Human kidney tissue in vitro is capable of modelling human genetic disease, whether by using patient-derived iPSC or by introducing mutations through genome editing techniques such as CRISPR/Cas9 or transcription activator-like effector nucleases (TALENS). The paucity of therapeutics for PKD, which accounts for 10% of ESKD, provides motivation for clinical-trials-in-a-dish using hPSC-derived PKD models. ARPKD, while less prevalent, may be a better model than ADPKD owing to its developmental phenotype. Alternatively, an ADPKD model could be used to identify mechanisms by which a dominant disease can engender a delayed age of onset, such as the second hit hypothesis. Future work to determine the directed differentiation protocol for terminal UB may provide a better tissue model for PKD, as cysts are generally concentrated in the collecting system. Coculture of UB and MM would represent an ideal model for studying congenital anomalies of the kidney and urinary tract, which often arises from failure of the reciprocal induction between the two tissues. Experiments using kidney tissue generated from patients with APOL1-associated focal segmental glomerulosclerosis may elucidate the concerted factors that transition carriers of the APOL1 risk variant to primary focal segmental glomerulosclerosis.
In the future, non-genetic kidney diseases may be modelled in stem cell-based human tissue. Severe or recurrent acute injury to hPSC-derived kidney tissue, possibly with fibrogenic cells and/or macrophage coculture, may recapitulate the pathophysiology leading to kidney fibrosis, a common hallmark of varied causes of progressive chronic kidney disease. Using kidney-on-a-chip techniques to generate perfusable glomerular structures, perfusion with isolated immune complexes from patients’ serum may permit modelling of lupus glomerulonephritis, cryoglobulinemia, post-streptococcal glomerulonephritis, and immunoglobulin A nephropathy. The application of anti-phospholipase-A2-receptor antibodies may simulate membranous nephropathy. Culturing organoids in a hypoxic incubator may model kidney ischaemia. Disease modelling in human tissue in vitro represents a formidable tool for drug screening, aiding the identification of novel therapies in human disease.
Regenerative medicine strategies employing existing methods for hPSC-derived kidney tissue face challenges related to the lack of a perfusable vascular tree and organised collecting system.Advances in biomedical engineering include the generation of stable vascular networks in 3D printed ECM-based hydrogels. The coculture of developing stem cell-based human kidney tissue with a pre-existing vascular network may generate a perfusable vascular tree. Determining the optimal timing along the directed differentiation protocol to conduct coculture and composition of the ECM-based hydrogel may facilitate glomerular vascularisation. Perfusion of glomerular capillary tufts in vitro may elicit a filtrate, for which sieving coefficients could be determined using dextran particles of differing size and charge. A collecting duct system has been previously generated using isolated mouse MM and UB in vitro. Using similar methods, the coculture of hPSC-derived MM and UB tissue may induce a common branched collecting duct system. Alternatively, the decellularisation-recellularisation technique represents a pre-formed scaffolding approach towards bioartificial kidney creation. One such approach involves dissociating multicellular hPSC-derived kidney organoids to single cells and seeding decellularised mammalian kidney via the renal artery, renal vein, and ureter. The influence of existing compartmentalised kidney ECM, and the retained growth factors, may facilitate cellular attachment and proliferation in proper compartments. The increasing need for novel forms of renal replacement therapy will surely lead to considerable advances towards a bioartificial kidney, as necessity breeds innovation.