Ethical approval statement: Ethical approval for this study was provided by an institutional review board, and consent to participate was obtained from the patient’s guardian with informed verbal consent.
Abstract
Pulmonary lung involvement in adult-onset Still’s disease (AOSD) can be classified into two categories: with or without acute respiratory distress syndrome. Interstitial fibrosis in AOSD is rare, occurring in less than 5% of cases. Here, the authors present a case of a 40-year-old male of Asian descent with a past history of hyperthyroidism who presented with fever, shortness of breath associated with cough, sore throat, diffuse arthralgias, pink-coloured rash, and hepatomegaly. Laboratory investigations revealed leukocytosis, abnormal liver function tests, negative antinuclear antibodies, and negative rheumatoid factor. Chest X-ray showed bilateral basal infiltrates, while high-resolution CT chest scan confirmed the diagnosis of interstitial lung disease in association with ASOD. This case suggested a direct association of AOSD and interstitial lung disease, but autoimmune pathogenesis is the only link synchronising both diseases and the exact mechanism of direct involvement is ambiguous.
INTRODUCTION
Adult-onset Still’s disease (AOSD) is an inflammatory disorder characterised by the classical triad of quotidian (daily) fever, arthralgia, and evanescent rash.1 In 1896, George Still2 first acknowledged this triad of quotidian fevers, evanescent rash, and arthritis in children, which was later termed juvenile inflammatory arthritis. In 1971, Bywaters3 described a series of adults who had similar characteristics to children with systemic juvenile idiopathic arthritis but failed to meet the criteria for classic rheumatoid arthritis (RA), which was later called AOSD. The prevalence of AOSD is 1.5 cases in 100,000–1,000,000 with a bimodal age distribution of 15–25 and 36–46 years of age.4
Countless infectious agents have been proposed in association with AOSD, including many viruses.5Chlamydia pneumoniae, Mycoplasma pneumoniae, Yersinia enterocolitica, and Borrelia burgdorferi are the known bacterial inducers.6 One case review also reported AOSD with Rocky Mountain spotted fever.7
AOSD can be divided into two distinctive phenotypes, i.e., a systemic pattern and articular pattern based on cytokine profile, clinical features, and laboratory investigation.8 Mechanisms for autoimmunity, such as activation of macrophage, inflammatory cytokines (e.g., IL-1, IL-6, IL-18, IFNδ, and TNFα), and genetic factors, along with infection play a role in the pathogenesis of AOSD as indicated in literature.9 Prognosis varies greatly according to the course of illness and particular conditions, from benign and restricted outcomes to chronic damaging polyarthritis and life-threatening events, occurring from visceral complications or diseases.10
CASE PRESENTATION
A 40-year-old male of Asian descent previously diagnosed with hyperthyroidism with a historyof recurrent respiratory tract infections reported to the authors with a complaint of high-grade fever >40 ⁰C, dyspnoea associated with cough, sore throat, and diffuse joint pains. The patienthad symptoms of generalised weakness, decreased appetite, joint pains, and clinically marked weight loss for the last year. When asked, the patient did not report any chest pain, syncope, haemoptysis, night sweats, active or passive smoking, or exposure to pets; additionally, they did not have any travel or contact history in the recent past, intravascular drug abuse, or any other active complaint. The patient was commenced empirically on intravenous levofloxacin, intravenous hydration with 0.9% normal saline, vitamin B-12, folate supplements, and ibuprofen for their fever, generalised weakness, and arthralgia.
The physical examination was unremarkable except for moderate anaemia; two enlarged lymph nodes in the right inguinal region; diffuse pink-coloured rash spreading over the neck and forearms, which disappeared with resolution of fever (Figure 1); Grade 1–2 synovitis in proximal and distal interphalangeal joints; reduced chest expansion in the lower chest with fine inspiratory crepitations from mid to lower chest bilaterally; and hepatomegaly. The laboratory outcomes were haemoglobin: 6.3 g/dL, mean corpuscular volume: 72 fL, total leukocyte count: 17×103 U/L, granulocyte count: 85%, platelets: 458×103 U/L, C-reactive protein: 78 mg/L, erythrocyte sedimentation rate: 125 mm/hour, lactate dehydrogenase: 2398 U/L, serum total bilirubin: 2.23 µmol/L, direct bilirubin: 1.40 µmol/L, alanine amino-transaminase: U/L, aspartate amino-transaminase: 107 U/L, alkaline phosphatase: 299 IU/L, gamma glutamyl transferase: 85 U/L, serum ferritin: 3,219 ng/dL; blood, sputum, and urine cultures were negative.
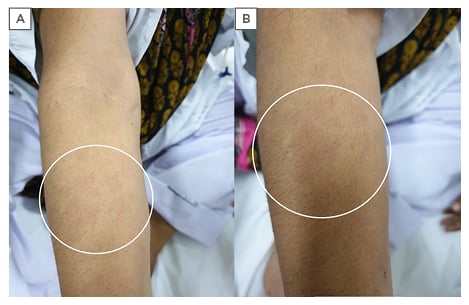
Figure 1: Rash over the right forearm. A) Rash disappears with fever settling; B) zoomed image to show the pink colour of the rash.
Viral markers and serologies were negative for influenza, pneumococcus, chlamydia, mycoplasma, and streptococcus. Chest X-ray showed bilateral basal infiltrates; a prior chest X-ray with no interstitial shadowing taken 1 year previous is shown for comparison (Figure 2). Pulmonary function tests showed a mild restrictive defect without obstruction with a forced vital capacity (FVC) of 2.87 L (predicted 65%), forced expiratory volume in 1 second of 2.21 L (predicted 61%), forced expiratory volume in 1 second /FVC of 77%, total lung capacity 4.30 L (predicted 59%), diffusing capacity for carbon monoxide of 25.15 mL/min/mmHg (corrected for haemoglobin 16.30 mL/min/mmHg), and FVC/diffusing capacity for carbon monoxide of 1.26. Antinuclear antibodies and rheumatoid factors were negative, and the rest of the labs were unremarkable. The differential considerations at this point included sarcoidosis, disseminated tuberculosis, AOSD, and low-grade lymphoma.
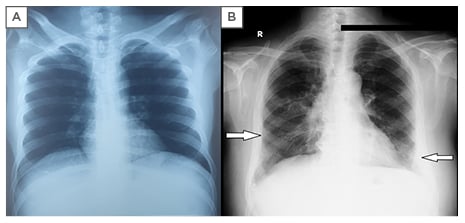
Figure 2: Chest X-ray (posteroanterior view).
A) Chest x-ray of 1 year prior; B) bilateral basal infiltrates.
The patient was scheduled for high-resolution CT (HRCT) scan of the chest, which showed irregular interlobular septal thickening with moderate bronchiectasis changes and subpleural reticulations in both lower lobes and right middle lobe with relative volume loss in lower lobes bilaterally, suggestive of interstitial pulmonary fibrosis with a probable pattern of usual interstitial pneumonitis without honey-combing, according to the 2018 American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and Latin American Thoracic Society (ALAT) classification system (Figure 3).11 The most plausible diagnosis made, based on clinical and laboratory findings, was interstitial lung disease (ILD) in association with AOSD. All of the nine diagnostic criteria of AOSD were fulfilled in the patient. A baseline X-ray chest of 1 year prior showed no interstitial changes, hence it was presumed that the patient might have chronic Still’s disease because the patient had articular complaints for the last year, which was later diagnosed during a flare and, therefore, its sequelae of ILD also presented late. During the hospital stay, the patient had intermittent spiking fever (range from 38.0 ⁰C to 40.1 ⁰C) with disappearing evanescent rash on the resolution of fever. The patient was given methylprednisolone initially in pulse dose (750 mg daily for 3 days), followed by maintenance dose prednisolone (15 mg per day) and mycophenolate mofetil (500 mg twice daily) was used later as a steroid-sparing agent for a short term, although no studies found suggesting its role except for complications like glomerulopathy, haemophagocytic lymphohistiocytosis, and lichen planus.12,13 In addition to empiric management, starting tacrolimus was planned if any flare-up of the disease occurred during the follow-up because there have been studies suggesting the role of tacrolimus in refractory Still’s disease.14,15 Subsequent follow-up in the ambulatory outpatient clinic setting was uneventful, hence tacrolimus was not used in the patient.
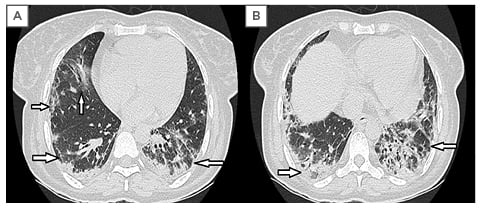
Figure 3: High-resolution CT scan of the chest.
A) Subpleural reticular densities with irregular interlobular septal thickening and relative volume loss in both lower lobes; B) fibrosis, traction bronchiectasis, and crowding of the involved bronchi, predominantly in the lower lobes. Signs are suggestive of interstitial lung fibrosis (marked with arrows).
DISCUSSION
High spiking fever and systemic derangements are the conventional presentations of AOSD.16 Typical rash, arthralgias and/or myalgias, lymphadenopathy, and derangements of liver function enzymes are appreciated. The fever exceeding 39 ⁰C is quotidian in nature, spiking in the evening.17 The characteristic rash is usually quoted as evanescent, pink coloured, and maculopapular in nature, targeting extremities and trunk, with sparing of the face. Non-specific histological findings are suggestive of dermal inflammation by lymphocytes and histiocytes with perivascular invasion.18 An overwhelming majority of patients (64–100%) are distressed with arthritis and arthralgias affecting knees, ankles, and wrists as commonly targeted joints, but arthritis and arthralgias of elbow, shoulder, and hips are also reported.19 Wrist manifestations depict the narrowing of joint space and ankylosis within 3 years of disease occurrence in untreated patients.20 Radiography is not usually productive in early detection of disease. Abnormal liver function tests are recorded in 75% of the patients with AOSD.21 Pericardial effusions and myocarditis are reported cardiac complications.22 Pleural effusions, fibrosis, and seldom acute respiratory distress syndrome (ARDS) constitutepulmonary involvement.23
Pulmonary associations with AOSD are rare, accounting for 12–35% for pleuritis and 0–27% for interstitial pneumonia.23 Pulmonary lung involvement (PLI) in AOSD can be classified into two categories: with acute respiratory distress syndrome or with other PLI.24 Interstitial fibrosis in AOSD is rare, occurring in less than 5% of cases.20 Unilateral or bilateral pleurisy is reported in 5–31% of cases. ILD is seen in 13% of cases, with transient radiological infiltrations,23 which are usually asymptomatic. A case of AOSD with organising pneumonia based on HRCT and transbronchial biopsy findings has also been reported.18 Proinflammatory cytokines, specifically IL-18, play a vital role in displaying pulmonary manifestations in AOSD. Acute intervals of the disease are characterised by peaking levels of IL-18 in serum and pulmonary tissues along with spiking levels of serum ferritin.16 Abnormalities of pulmonary functions described above can be recognised either in the presence or absence of symptoms.17 ILD is diagnosed on the basis of a HRCT scan of the chest.21 The restrictive ventilatory pattern in AOSD is said to be responsive to combined steroid and aspirin therapy.22Histological lung features in AOSD include non-specific interstitial fibrosis, acute alveolitis with thickened alveolar walls, hyperplasia of Type II pneumocytes, hyaline membrane formation, and l ymphocytic infiltration.23
There are many diagnostic criteria used for the definitive diagnosis of AOSD, with Yamaguchi’s being most widely used.25 Fever, arthralgia, typical rash, and leukocytosis are major criteria for Yamaguchi, while sore throat, lymphadenopathy, hepatosplenomegaly, abnormal liver function tests, negative antinuclear antibodies, and negative rheumatoid factor are minor criteria. A minimum of five criteria, with at least two being major, is required for the diagnosis of AOSD. This present patient fulfilled all the major and minor criteria required for the diagnosis.
Treatment of AOSD and PLI–AOSD remains empirical.26 Non-steroidal anti-inflammatory drugs, corticosteroids, immunosuppressive drugs (methotrexate, leflunomide, gold, azathioprine, cyclosporin A, cyclophosphamide), and intravenous gamma-globulin constitute an extensive treatment modality for AOSD. The recent productive trials of biological agents (anti-TNF, anti-IL-1, and anti-IL-6), usually combined with conventional immunosuppressive drugs, have been shown to be effective.27 Corticosteroids are the first-line treatment for AOSD, without taking the clinical presentation into account. Moreover, corticosteroids are more effective for systemic symptoms compared to articular ones. Methotrexate remins the first-line, steroid-sparing therapy in AOSD, while anakinra (IL-1 antagonist), and tocilizumab (anti-IL-6 receptor) are possible alternatives. Plasma exchange and intravenous Ig are other treatment options in refractory AOSD patients.2,27
This case presented a patient with fever, shortness of breath, pink-coloured rash, and hepatomegaly, and the diagnosis of ILD was met in association with ASOD. The limitation of the study is a lack of histological evidence of the disease process because bronchoscopy could be performed due to consent not given by the patient.
CONCLUSION
This case describes a direct association of AOSD with ILD, although early in the course of ILD, many patients are misdiagnosed with recurrent pulmonary infections, when in fact, they have early ILD.28 Therefore, in a patient who was diagnosed with AOSD and complaining of respiratory problems, ILD should be ruled out beforehand so that the management of AOSD can be adjusted in that particular patient. Moreover, the authors assume that both diseases can interact with each other, but the mechanism of direct association is still unknown.






