Abstract
Lung transplantation is increasingly used as a last resort treatment for end-stage chronic lung diseases. In the last decade, important improvements have been noted in terms of surgical techniques and perioperative and postoperative management, as well as greater acceptance of older recipients. However, the most drastic changes are observed in recipient and donor profiles due to advances in the treatment of chronic lung diseases. Such advances include novel patient stratification systems, such as the concept of progressive fibrosing interstitial lung diseases in pulmonary fibrosis; novel findings regarding etiology of disease, such as telomere length in pulmonary fibrosis; novel pharmaceutical treatment options, such as CFTR modulators in cystic fibrosis; and novel interventions, such as lung volume reduction in chronic obstructive pulmonary disease, and endarterectomy and balloon angioplasty in chronic thromboembolic pulmonary hypertension. In this review, the authors discuss the latest developments in the management of chronic lung diseases and how this affects recipient profiles and lung transplant management. Finally, the authors point out the uncertainties and knowledge gaps in these developments regarding transplant management.
Key Points
1. Lung transplantation is a valuable treatment option for patients with end-stage chronic lung disease. Recipient and donor profiles have changed significantly over the years due to advances in the treatment of chronic lung diseases, such as novel patient stratification systems, novel findings regarding disease etiology, novel pharmaceutical treatment options, and novel interventions.2. Pulmonary fibrosis is now the most common indication for lung transplantation worldwide. The impact of telomeropathies on post-transplant outcomes needs to be further assessed. This should include adequate risk assessments for post-transplant complications related to the type of underlying telomeropathy, as well as guidance on how immunosuppressive regimens should be optimized in these patients.
3. Chronic obstructive pulmonary disease also remains a common indication for lung transplantation. As the role of lung volume reduction in the treatment of emphysema continues to expand, further studies assessing the peri-transplant risks and benefits of prior lung volume reduction, as well as its use as a bridge to transplant, should be conducted.
INTRODUCTION
It is clear that the landscape of clinical lung transplantation has noticeably changed since its introduction in the 1960s. Surgical techniques, as well as perioperative and postoperative patient care, have improved significantly, contributing to better short- and long-term outcomes.1 Changes are also evident in the patient population. Parallel to that of the general population, the average age of both lung donors and recipients continues to rise globally. In Europe, donors over the age of 65 are becoming increasingly common, while in the United States, the number of lung transplants in recipients over 65 has doubled over the past decade, now accounting for over 30% of transplant activity.2-4
The profile of lung transplant patients is not only changing in terms of age, but in other factors as well. The advancement of various medical therapies (e.g., antifibrotic agents) and surgical interventions (e.g., lung volume reduction) has influenced the need and timing of lung transplantation. In addition, more patients with comorbidities and technically complex cases, such as those with prior interventions, are now being considered for transplantation. Still, substantial variation remains in clinical practices across transplant centers and regions worldwide.
In this review, the authors focus on changes in the patient population and timing of lung transplantation from a disease-specific perspective for the major transplant indication groups. Rarer indications for lung transplantation, including acute respiratory distress syndrome,5 pulmonary chronic graft-versus-host disease,6 and re-transplantation,7 are described elsewhere.
LUNG TRANSPLANTATION FOR PULMONARY FIBROSIS
Pulmonary fibrosis comprises a group of chronic fibrotic interstitial lung diseases (ILD) that carry a substantial risk of progressive respiratory failure and death, despite optimal medical treatment.8
Main Indication for Lung Transplantation
Indications for lung transplantation have evolved, and ILD has surpassed chronic obstructive pulmonary disease (COPD) as the leading indication for lung transplantation worldwide.3 In addition to increased awareness and diagnosis of ILDs, this is also due to the implementation of the Lung Allocation Score (LAS), a numerical score that prioritizes patients based on medical urgency and likelihood of successful transplant outcomes.9 Since the implementation of LAS, more high-priority patients have received transplants, leading to changes in recipient profiles. As reflected by LAS values, disease severity has increased and waiting times have decreased for patients with ILD.9 The same prioritization principles apply under the newer Composite Allocation Score (CAS) system in the US.
Moreover, the tendency to transplant older candidates has shifted transplant indications as well, as ILDs tend to occur later in life than COPD. According to 2017 registry data from the International Society for Heart and Lung Transplantation (ISHLT), ILD accounted for 40% of transplant indications, followed by COPD (26%), and cystic fibrosis (CF) at 13%.3 The number of transplants for idiopathic pulmonary fibrosis (IPF) has risen substantially in recent years, particularly in the US.10
At the authors’ large-volume center, which has performed >1,500 lung transplants since the introduction of its program in 1991, transplant indications have similarly shifted. While the center continues to primarily transplant patients with COPD, there has been a noticeable rise in the number of ILD transplants, whereas CF transplant activity has dropped to nearly zero in recent years (see Figure 1).
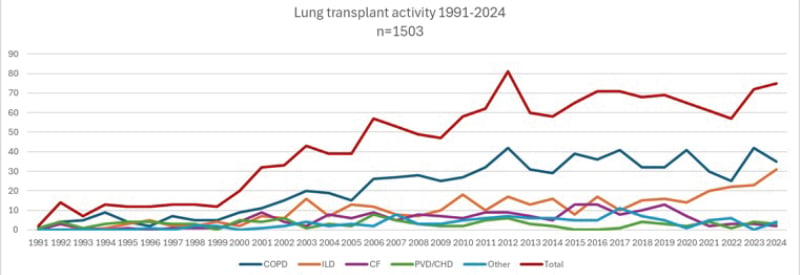
Figure 1: Changes in lung transplant indication.
Overview of lung transplant activity at the Leuven lung transplant program, a high-volume center that has performed over 1,500 transplants from the introduction of its program in 1991 to 2024. Although the transplant activity for COPD has remained relatively stable, a clear increase in the number of ILD transplants has been observed, while CF lung transplant activity has almost completely decreased to zero in recent years.
CF: cystic fibrosis; CHD: congenital heart disease; ILD: interstitial lung disease; PVD: pulmonary vascular disease.
The Concept of Progressive Fibrosing ILD
The most fundamental paradigm shift in fibrosing ILD management is the advent of the progressive fibrosing ILD (PF-ILD) concept, acknowledging that subgroups of patients with a non-IPF fibrosing ILD diagnosis behave similarly compared to IPF in terms of pulmonary function decline,11 survival,12 and treatment response to antifibrotics.13 IPF remains the most common type of ILD for lung transplantation, but other PF-ILDs, such as fibrotic hypersensitivity pneumonitis and idiopathic non-specific interstitial pneumonia, have increased as indications for transplantation.3,14 Interestingly, the PF-ILD criteria are already woven into the ISHLT guidelines on transplant candidacy for referral and listing. Patients with IPF and patients with progressive pulmonary fibrosis (PPF) are both recommended to be referred for lung transplantation intake and evaluation.
Impact of Telomere Disorders
Currently, it is recognized that genetic variants linked to short telomeres are prevalent in patients with pulmonary fibrosis, even among those without a known family history of ILD. Besides pulmonary fibrosis, telomere shortening and related genetic mutations are associated with disorders in other organ systems, including myelodysplasia, cirrhosis, and hematological and solid organ malignancies.8 Short telomere length (<10th percentile), often caused by specific telomere-related mutations (e.g., TERC or TERT), has also been connected to worse post-transplant outcomes and may increase the risk of hematological complications, cytomegalovirus-related complications, and malignancy. Studies on the impact of short telomere length and chronic lung allograft dysfunction have shown conflicting results.15-20 Routine pre-transplant testing for telomere length and/or associated mutations is performed in candidates with ILD in some centers. Currently, there is no consensus on the post-transplant (immunosuppressive) management of patients with telomere disorders, but an ISHLT consensus document is being developed (Table 1).
Pre-transplant Immunosuppression
Medical therapy of pulmonary fibrosis is based on the specific underlying diagnosis and disease behavior, and may include immunosuppression in non-IPF entities. Any potential benefit of ongoing immunosuppression at a fibrotic disease stage must be weighed carefully against its risks.8 While deleterious effects of immunosuppression have been acknowledged for years in IPF, evidence is emerging that this treatment in patients with non-IPF fibrotic ILD and short telomere length portends worse survival,21 and its use should be stringently evaluated. In general, it is best to limit immunosuppressants to the lowest effective dose while a patient is listed for transplantation. In particular, chronic, high-dose corticosteroids may increase the risk of wound healing complications and anastomotic dehiscence, and should be avoided if possible.14
Impact of Antifibrotic Agents on the Need and Timing of Lung Transplantation
Two antifibrotic agents, pirfenidone and nintedanib, are currently used in patients with pulmonary fibrosis. Their availability has altered the previously known clinical disease course by attenuating functional decline and disease progression and potentially increasing survival.14,22-26 Despite these positive findings, however, antifibrotics merely slow down the disease and do not stabilize or improve pulmonary function.22,24 Hence, progressive and ultimately fatal disease remains the long-term outcome for both IPF and PPF cases, for which lung transplantation remains the only definitive treatment option in well-selected patients. As such, the use of antifibrotic agents may postpone the need for a transplant. This could lead to a greater demand for transplants at older ages, which could present societal and ethical challenges while placing pressure on centers that apply strict age limits.
IPF has been associated with cardiovascular and other comorbidities,27,28 and aging further increases these risks. Age remains a controversial selection criterion for lung transplantation. Nevertheless, the recipient population is aging, partly due to an increasing number of older patients with pulmonary fibrosis receiving transplants. Frailty assessment is essential, especially in older patients, to distinguish chronological age from biological and functional age.
Importantly, most studies show that antifibrotics are generally safe up to the time of transplant, with no significant effect on suture complications, bleeding, or reintervention rates.29
When To Transplant: Risk of Exacerbation
The ISHLT has established disease-specific recommendations to help determine the appropriate timing for referral and listing of patients with pulmonary fibrosis.30 In general, two-thirds of both PPF and IPF cases experience a forced vital capacity decline of >5% in one year, and almost half of patients experience a >10% decline.12,31 Still, the clinical course of pulmonary fibrosis is variable. Some patients remain stable for extended periods, while others deteriorate rapidly or incrementally. As such, determining the ideal time to place a patient on the waiting list and proceed with the transplant is not easy and largely depends on the individual’s disease trajectory. Other risk factors, such as secondary pulmonary hypertension and concurrent emphysema, should also be taken into account.32 In patients with combined pulmonary fibrosis and emphysema, diffusion capacity is typically severely impaired, whereas airflow rates and lung volumes are relatively preserved. The latter may contribute to underestimation of disease severity and complicate monitoring of disease trajectory by lung function, as changes in forced vital capacity are not reliable indicators of disease progression in this patient group.33
The unpredictable nature and potentially severe consequences of ILD exacerbations must be recognized, and in some cases, patients who were not previously considered or fully assessed for lung transplantation may need to be urgently evaluated.14
Unique Features Related to Pulmonary Fibrosis
Several unique characteristics in patients with pulmonary fibrosis may influence lung transplant candidacy, as well as pre- and post-transplant risks and management strategies. For instance, the importance of telomere biology disorders, extrapulmonary disease manifestations, and allosensitization in patients with connective tissue disease should be considered.14,34
Historically, patients with connective tissue disease-related ILD were considered suboptimal candidates for lung transplantation due to multi-organ disease manifestations and surgical risk. Severe esophageal dysmotility and gastroesophageal reflux are of particular concern, along with the need for cardiovascular evaluation and assessment of gastrointestinal, renal, musculoskeletal, and other organ systems.8,34 Importantly, no difference in survival or allograft function was noted in carefully selected patients compared with patients transplanted for other indications.8,14 In the context of extrapulmonary disease manifestations, it is also necessary to be vigilant for the possibility of cardiac involvement in sarcoidosis.
Given that several post-transplant complications are unique to this patient population, multidisciplinary management by experts familiar with telomere disorders and infectious, hematological, oncological, and cardiac complications may be necessary to increase the likelihood of better outcomes.8,14
LUNG TRANSPLANTATION FOR COPD
COPD has historically been the most common indication for lung transplantation. Although ILD has surpassed COPD as the most common indication globally, COPD remains an important indication too, despite medical and surgical advancements in its treatment.3
Impact of Lung Volume Reduction Techniques on the Need for Lung Transplantation
Endoscopic and surgical lung volume reduction (ELVR and LVRS) are increasingly used therapeutic options for well-selected patients with emphysema who have not yet progressed to end-stage lung disease.35 LVRS, in which diseased areas of lung parenchyma are resected, may lead to improved exercise capacity, quality of life, and survival in carefully selected patients.36,37 Insertion of endobronchial valves is the most common technique for ELVR, and has positive effects on lung function, exercise tolerance, and quality of life.38 Despite these clinical benefits, pulmonary function may decline further in some of these patients, ultimately requiring a lung transplant.39 Both LVRS and ELVR can be considered bridging strategies, as they do not prohibit future lung transplantation.35 Indeed, some centers are now using LVRS to delay the need for transplantation, which could potentially improve overall survival, or to stabilize and enhance a candidate’s pulmonary and general status prior to transplantation.
Impact of Prior LVRS on Transplant Outcomes
LVRS before lung transplantation has long been controversial, with outcomes still not entirely understood. Single-center studies have reported conflicting results.39 Some of them observed increased perioperative risks, such as graft failure, increased need for blood transfusion, extracorporeal membrane oxygenation, and empyema, as well as longer hospital stays after LVRS and higher wound infection rates following ELVR.39,40 However, neither short- nor long-term survival was negatively affected.40
These risks may stem from the technical challenges of transplanting patients who have previously undergone LVRS, including pleural adhesions, which can increase operative time, bleeding complications, phrenic nerve injury, and transfusion needs.39,40 Interestingly, the multicenter study by Krishnan et al.39 noted that the proportion of lung transplants with prior LVRS ranged from 0–11% per center, suggesting that outcomes may benefit from specialized expertise and institutional volume, other technically complex procedures. Theoretically, it could also be the case that patients requiring lung transplantation after LVRS represent a more severe phenotype of COPD.39
Thus, while LVRS prior to lung transplantation does not seem to affect post-transplant survival, it may increase perioperative risk.39,40 Therefore, such patients require thorough pre-transplant evaluation.39 As the role of lung volume reduction in the treatment of COPD continues to expand, further studies assessing the risks and benefits of prior LVRS/ELVR and their use as a bridge to transplant should be conducted (Table 1). ELVR is a less invasive approach and has the potential to reduce some of the risks associated with LVRS that could complicate subsequent lung transplantation.39

Table 1: Uncertainties and knowledge gaps in (pre-)lung transplant management due to advances in chronic lung disease care.
CF: cystic fibrosis; ELVR: endoscopic lung volume reduction; LAS: lung allocation score; LVRS: lung volume reduction surgery; PAH: pulmonary arterial hypertension; PF: pulmonary fibrosis; REVEAL: Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Impact of Targeting Type 2 Inflammation in COPD
It is known that COPD is a heterogeneous disease, and that Type 2 inflammation occurs in 20–40% of COPD cases.41 In such cases, targeting Type 2 inflammation can reduce acute exacerbation rates by up to 30%.42,43 As these treatment options have not yet been routinely used, their impact on pulmonary function decline, transplant indication, and timing is not yet fully elucidated. However, it is clear that these therapies, which significantly lower COPD exacerbation rates, will have a profound impact on the COPD transplant landscape.
LUNG TRANSPLANTATION FOR CYSTIC FIBROSIS
CF is the most common autosomal recessive disorder in White people, and it is caused by biallelic mutations in the gene encoding for the CFTR protein.44 Until recently, it was the third most common indication for lung transplantation.3 Nowadays, the number of patients referred for and transplanted due to CF continues to decline drastically as a result of multifaceted symptomatic management and new disease-modifying drugs, with favorable pre-transplant evolution and increased life expectancy.44
Impact of CFTR Modulators on the Need for Lung Transplantation
CF is a multisystem disorder, with CF lung disease causing the majority of morbidity and mortality. Lung transplantation is the ultimate treatment option for patients with progressive, advanced lung disease, with superior long-term survival outcomes compared with other primary underlying diseases, which is associated in part with younger age at the time of transplant.3,45 However, the introduction of CFTR modulators, which target the underlying defect, has profoundly transformed CF management. These modulators have substantially impacted the disease course of many patients with CF by significantly improving lung function, quality of life, and predicted survival rates.44
These drugs are highly effective and, as a result, there has been a marked reduction in the number of patients referred for and transplanted due to CF in recent years.2,46 Although an increasing amount of data confirms the long-term efficacy of these drugs, patients with already advanced lung disease might still need a lung transplant later in the course of their disease.47-52 This may also affect the patient profile, with a potentially increased perioperative risk profile due to age-related comorbidities (cardiovascular, renal disease) or CF-related liver disease.53
With CFTR modulators now being introduced at a younger age, CF lung disease will likely be prevented in the majority of patients. There remains a small subgroup of patients with CF who have rare CFTR mutations and are currently ineligible for or demonstrate a heterogeneous response to CFTR modulator therapy.54 Although generally well tolerated, there is also a small group of patients with CF who have to discontinue their CFTR modulators due to side effects.55 However, as the CF drug discovery pipeline continues to expand with various nucleic-acid-based strategies in Phase I and II clinical studies,56 it seems reasonable to expect that CF will become an increasingly rarer indication for lung transplantation.
Lung transplant referral and listing recommendations have not yet incorporated the impact of the new CFTR therapeutics. Currently recommended thresholds for transplant referral and listing require reassessment, especially as more data become available on the long-term effects of CFTR modulators and disease progression in patients with advanced lung disease.30,44
Is There a Role for CFTR Modulators After Lung Transplantation?
Patients with CF who are undergoing lung transplantation require specialized post-transplant care due to the multisystem nature of CF and its unique comorbidities. The Cystic Fibrosis Foundation (Maryland, USA) consensus statement provides guidance for post-transplant management.57 Because CFTR modulators can also have a positive effect on other tissues and organs where CFTR is expressed, interest is emerging in the continuation or initiation of CFTR modulators after lung transplantation.
Infections remain a significant issue post-transplant and are attributed to chronic immunosuppression as well as pre-transplant colonization with typical CF pathogens, including the colonization of the upper respiratory tract in individuals with CF-related sinus disease. Organisms such as Pseudomonas aeruginosa, along with gastroesophageal reflux disease and (micro)aspiration, are associated with higher chronic lung allograft dysfunction (CLAD) risk. These factors provide some justification for the post-transplant use of CFTR modulators to mitigate extrapulmonary inflammation, which could influence CLAD development and/or progression, and improve quality of life.58
Although post-transplant data are scarce, CFTR modulators have shown benefits regarding extrapulmonary disease manifestations in non-transplant patients, improving chronic rhinosinusitis, gastrointestinal disease, diabetes, and possibly BMI and bone health.44,58 A Dutch multicenter study on lung transplant recipients with CF found that elexacaftor/tezacaftor/ivacaftor improved health-related quality of life, chronic rhinosinusitis, and gastrointestinal symptoms, though not BMI or HbA1c.59 Post-transplant drug monitoring is needed given the potential drug–drug interactions that can occur (e.g., increased calcineurin inhibitor levels), as CFTR modulators are metabolized through cytochrome P450 enzymes.44,58
There is a potential role for CFTR modulators after lung transplantation, as extrapulmonary CF comorbidities remain a considerable issue in some individuals.44 This was also supported by a patient survey, which showed that over 80% of respondents prioritized treating extrapulmonary symptoms, and 90% wanted more information on the post-transplant use of CFTR modulators. However, their high cost warrants further studies to better assess their risks, benefits, and potential impact on key outcomes such as quality of life and CLAD.58
LUNG TRANSPLANTATION FOR PULMONARY VASCULAR DISEASE
Patients with pulmonary vascular disease, mainly consisting of group 1 pulmonary arterial hypertension (PAH), group 4 chronic thromboembolic pulmonary hypertension (CTEPH), and group 5 pulmonary hypertension with unclear and/or multifactorial mechanisms, comprise approximately 5% of all lung transplants. 3 Therapeutic advances, including medical therapies and interventions such as pulmonary endarterectomy and pulmonary artery balloon angioplasty, have greatly improved the prognostic implications of pulmonary hypertension and led to a marked change in timing for referral and listing of patients.32,60 Additionally, improved cardiovascular surgical approaches in patients with congenital heart defects have drastically reduced the prevalence of Eisenmenger’s syndrome later in life as an indication for (heart-)lung transplantation.
Impact of Medical Treatment for PAH on the Need for a Transplant
Several types of drugs are available for the treatment of PAH, depending on the presence or absence of vasoreactivity and the results of risk stratification. Importantly, all of these drugs act in a non-permanent manner by inducing temporary vasodilation and reduction in pulmonary vascular resistance. As such, these drugs must be continued to maintain a beneficial effect on disease progression.61,62
Despite these medical options, survival remains poor, and lung transplantation remains the definitive treatment for selected patients with disease that is refractory to, or progressing despite, optimal therapy. Although revisions to the LAS have attempted to appropriately reflect disease severity and have improved the likelihood of transplantation for listed patients with PAH, it still does not fully reflect waitlist mortality, thus disadvantaging this population.30 Many patients with severe PAH receive a lung transplant after high-urgency listing due to life-threatening clinical deterioration.63 This also illustrates the difficulty in predicting clinical decline in patients with PAH.61
Lung transplantation for PAH is a complex procedure that requires a highly experienced team and access to circulatory support to achieve good post-transplant outcomes. Among lung transplant patients, patients with PAH have the highest perioperative mortality rate, although a favorable evolution has been observed due to improved surgical techniques; intraoperative management, with the implementation of extracorporeal membrane oxygenation; and prevention and management of primary graft dysfunction.60
Finally, more data are needed on post-transplant outcomes and timing of drug cessation regarding the recently approved PAH drug, sotatercept, a growth differentiation factor inhibitor that has been associated with increased risk of bleeding.64
Impact of Surgical Treatment for CTEPH: Is There Still Room for Transplantation?
CTEPH is characterized by the obstruction of proximal pulmonary arteries by fibrothrombotic material, along with microvasculopathy similar to that seen in PAH. In addition to lifelong anticoagulation, treatment options for CTEPH vary based on the patient’s profile and include: pulmonary endarterectomy; balloon pulmonary angioplasty, which is used for inoperable patients and patients with residual disease after pulmonary endarterectomy; and/or medical therapy.61 Due to these advancements in medical and surgical treatments, as well as the use of a multimodal approach,65,66 lung transplantation is now seldom considered for this patient group.3,61 Nevertheless, it may still be considered in well-selected patients with persistent or recurrent pulmonary hypertension, or in patients with very distal disease not amenable to other treatments.67 Notably, due to the well-developed bronchial collateral circulation, lung transplantation in these patients can be technically difficult and may carry a substantial risk of bleeding complications.61
CONCLUSION
The demographic profile of lung transplant candidates is shifting, with an increasing number of older patients and patients with complex comorbidities being considered for transplantation. Pulmonary fibrosis and COPD continue to be important transplant indications, although the role of new therapies, such as antifibrotic agents in pulmonary fibrosis, lung volume reduction techniques in COPD, CFTR modulators in CF, and multimodal vascular management in PAH, is reshaping the timing and need for transplants. Overall, the evolving landscape of lung transplantation presents both challenges and opportunities, requiring continued refinement of patient selection, timing, and management strategies to ensure optimal outcomes.