Abstract
The immune system constitutes a critical mechanism of the human body to preserve health and mitigate disease. In the lung, immunity is seen as a critical driver in many respiratory diseases, in particular in those characterised by aberrant inflammation, such as chronic obstructive pulmonary disease, fibrosis, and asthma. In this review, the specialised set of immune cells and lung tissue-specific regulators, including key cytokines such as granulocyte-macrophage colony-stimulating factor and transforming growth factor β, that control immune responses in the respiratory tract will be discussed. Furthermore, the current understanding of the impact of key environmental components such as the role of oxygen and lung microbiota on lung immunity will be highlighted. The goal is to identify the unique aspects of lung immune biology to facilitate insights into the aetiology of common lung inflammatory diseases and to provide the basis for a deeper mechanistic understanding of the underlying immune processes. Finally, key future avenues of research such as using more comprehensive quantitative approaches for elucidating molecular disease mechanisms as well as the potential to exploit tissue-specific regulators of immunity for therapy of lung inflammatory disorders will be discussed.
INTRODUCTION
As the mediator of gas exchange, the lung is an essential organ for mammalian survival and has thus evolved a diverse and intricate set of defence mechanisms to deal with challenges and still maintain its crucial functions. It has the second largest surface area of all human tissues and therefore has strong and constant exposure to the environment, including inhaled pathogens, allergens, particles, as well as resident micro-organisms. Similar to every organ system, the lung has a unique set of immune cells which, together with structural cells such as epithelial cells, form an interconnected network that orchestrates lung immunity. These cells include innate immune cells such as lung resident dendritic cells (DC), alveolar macrophages (AM), interstitial macrophages (reviewed in more detail here1), as well as basophils and mast cells. Both lung macrophages and DC together form a first line of defence against pathogen invasion while at the same time inducing some level of tolerance to prevent unnecessary inflammation. In addition, the lung also contains a high number of Type 2 innate lymphoid cells (ILC2) as well as other innate-like lymphocytes such as γδ T cells which play important roles in mediating lung immunity in health and disease. In recent years, the immune functions of various nonhaematopoietic cells as first sensors and transmitters of immune-relevant signals have also become apparent. While many of the underlying molecular mechanisms remain unclear and are being intensely investigated, all together the respiratory immune system reacts to internal and external challenges, promoting tissue homeostasis and repair but also contributing to acute and chronic pulmonary disease. Indeed, the lung has several intrinsic features such as high levels of oxygen that have a profound impact on the configuration of its immune system and its activity in respiratory illnesses. In this review, the current knowledge of how tissue immunity in the lung is regulated by these unique features including key host and environmental factors and how this orchestrates lung immune responses in health and disease will be discussed.
KEY ENVIRONMENTAL FACTORS REGULATING IMMUNITY IN THE LUNG
Oxygen
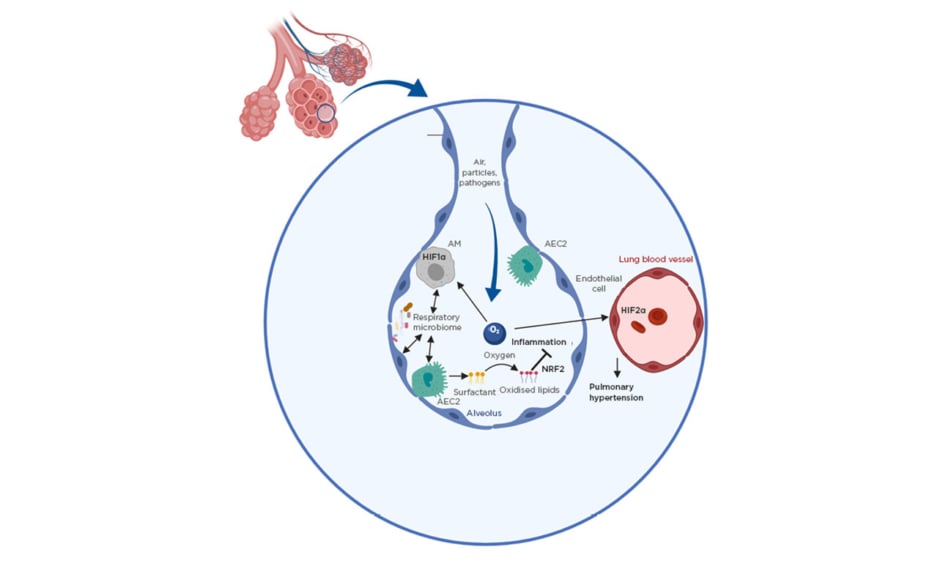
Because the function of the lung is to mediate gas exchange, the organ represents a unique environment with very high levels of oxygen compared to other tissues. This has a direct impact on lung immunity and disease (Figure 1). Firstly, transcriptional regulators that have activity directly linked to oxygen levels, in particular the hypoxia-inducible factor (HIF) family, play a key role in regulating lung disease in different contexts.2 HIF have been shown to be particularly important in regulating the development of pulmonary hypertension (PH) where local differences in oxygen levels can lead to activation of HIF1α which in turn promotes remodelling of the lung vasculature by inducing smooth muscle cell thickening.2 Furthermore, HIF2α induces pathological changes in endothelial cells in this context.3 It is now increasingly understood that immune cells play a key role in PH pathogenesis as HIF1α was found to promote disease in a myeloid cell intrinsic manner.4 Furthermore, there is evidence that the key regulators of HIF1α activity von Hippel-Lindau protein and prolyl-4-hydroxylase domain 3 directly regulate AM development and function.5,6 Furthermore, HIF1α activation in myeloid cells was found to promote asthma.7,8 A potentially detrimental role of HIF1α in promoting lung inflammation is becoming apparent in sarcoidosis in which it regulates inflammatory cytokines IL-1β and IL-17,9 and in chronic obstructive pulmonary disease (COPD), serum HIF1α is emerging as a useful biomarker for tracking disease progression.10 Conversely, while its role in lung infection is largely unclear, HIF1α has been shown to promote survival in chronic tuberculosis infection.11 More research is warranted in understanding its role in immunity to other pathogens such as respiratory viruses.
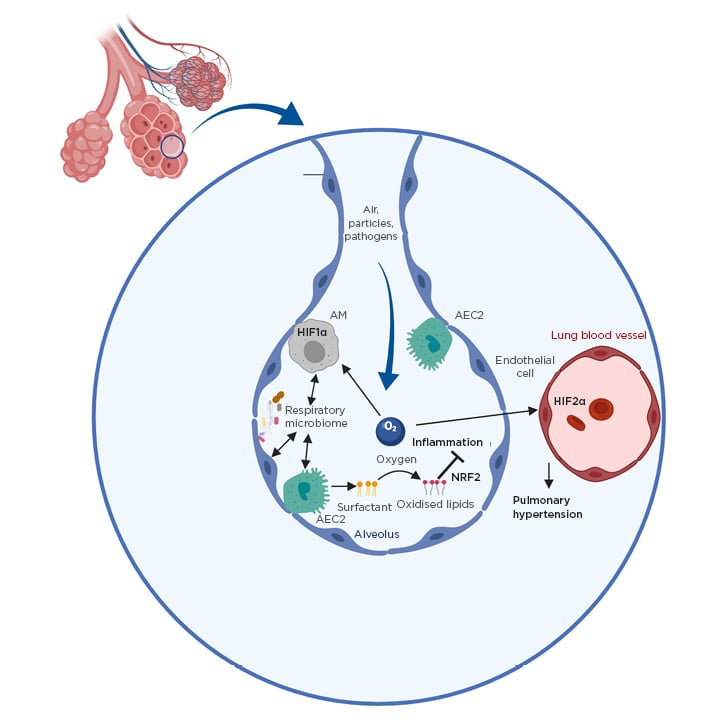
Figure 1: Environmental factors as important regulators of lung immunity.
Shown are key environmental factors controlling lung immunity. The roles of oxygen and the respiratory microbiota are depicted.
AEC2: Type II alveolar epithelial cell; AM: alveolar macrophage; HIF1/2α: hypoxia-inducible factor 1/2α; NRF2: nuclear factor erythroid 2-related factor 2.
Apart from directly regulating some host transcription factors, high oxygen levels in the lung oxidise the surfactant lipids continuously produced by Type II alveolar epithelial cells to facilitate gas exchange. In general, oxidised phospholipids are key regulators in inflammation, but are of particular importance in the lung because of the high availability of oxygen. Several mechanisms exist to control their levels, especially uptake and degradation by macrophages via scavenger receptors.12 Oxidised phospholipids were shown to negatively impact AM function13 but promote tissue repair after acute lung injury via activation of key transcription factor nuclear factor erythroid 2–related factor 2.14,15 Despite their high abundance in the lung, their role in many respiratory diseases is still completely unknown and more work is necessary to understand the importance of individual oxidised lipid species on lung inflammation.
Lung Microbiota
The lower respiratory tract harbours a unique microbiota, distinct in composition from other body sites such as the skin or the intestine.16 Indeed, the microbial composition in chronic lung disease is profoundly changed in conditions such as pulmonary fibrosis17 or COPD.18 While the functional impact of the respiratory microbiota on lung immunity is still unclear, bacteria are found in close proximity to lung epithelial cells,19suggesting an intimate relationship. Furthermore, there is emerging evidence that the microbiome in general has a broad impact on lung immunity, including influencing susceptibility to infection and development of asthma.20
While mechanistic insights are still largely lacking, there is some evidence that diet-induced changes in the intestinal microbiota lead to production of microbial metabolites such as short-chain fatty acids which in turn, via the circulatory system, control the development of allergic disease in the lung.21
Furthermore, such mediators can also control immunity to influenza virus infection by promoting antiviral T-cell responses,22 which is critical for viral clearance and recovery from infection. While in some cases beneficial, the microbiota was also shown to exacerbate the development of chronic lung inflammation in models of COPD23 as well as fibrosis.24 Indeed, more evidence is emerging that, not just in experimental settings but also in humans, the microbiota directly contributes to disease development or exacerbation.25For example, this includes lung fibrosis for which lung bacterial burden in patients was shown to correlate with disease progression.25 More research is warranted to investigate the underlying molecular mechanisms of microbiota–host crosstalk in the lung and how it contributes to pulmonary disease.
KEY HOST ORCHESTRATORS DETERMINING TISSUE-SPECIFIC IMMUNE RESPONSES
Several key host regulators of lung immunity have been discovered in recent years and how these factors regulate lung inflammation is displayed in Table 1.
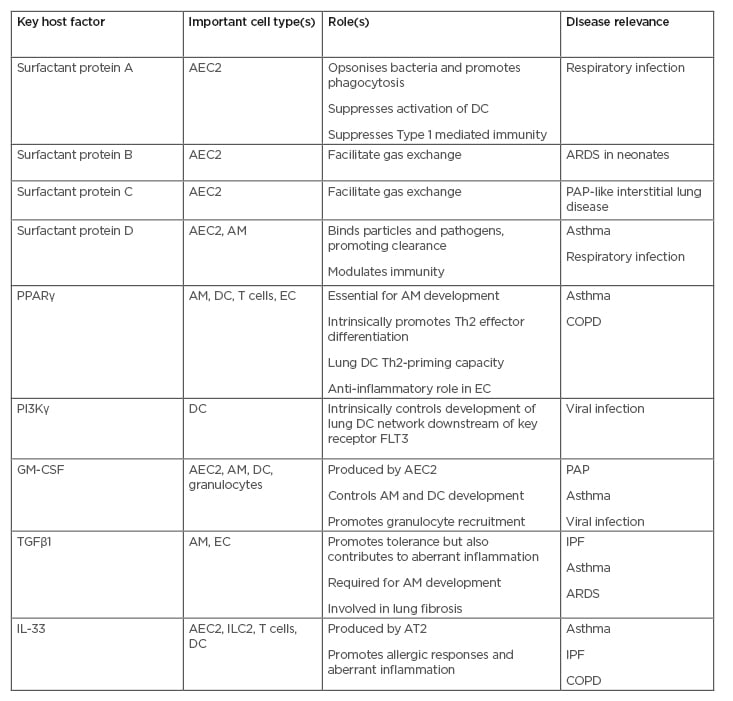
Table 1: Important host factors in lung immunity and their role in lung disease.
The roles of key host factors controlling tissue-specific lung immunity in the steady-state and in inflammation are shown in the table.
AEC2: Type 2 alveolar epithelial cell; AM: alveolar macrophage; ARDS: acute respiratory distress syndrome; COPD: chronic obstructive pulmonary disease; DC: dendritic cell; EC: epithelial cell; FLT3: FMS-like tyrosine kinase 3; GM-CSF: granulocyte-macrophage colony-stimulating factor; IPF: idiopathic pulmonary fibrosis; PAP: pulmonary alveolar proteinosis; PPARγ: peroxisome-proliferator activated receptor γ; Th: T helper.
Alveolar Surfactant
A particular key component in the alveolar microenvironment are surfactants, which are composed of a large variety of lipids such as phosphatidylcholine as well as several key proteins, notably the four surfactant proteins (SP) SP-A, SP-B, SP-C, and SP-D found in the alveolar space (Figure 2). By continuously producing Type II alveolar epithelial cells, the main purpose of the surfactant is to lower surface tension of the alveoli and thereby facilitate gas exchange. SP-B and SP-C are critical for maintaining this basic lung function. Congenital deficiency for SP-B, for example, is associated with severe respiratory distress syndrome in neonates.26 Furthermore, for mutations in the gene encoding for SP-C, severe interstitial lung disease and in some cases a pulmonary alveolar proteinosis-like phenotype has been described.27 However, in addition to these important roles in maintaining basic lung function, several key immunomodulatory properties have been described for SP-A and SP-D. When bacteria and other respiratory pathogens, as well as allergens and particles, enter the alveolar space, SP-D was shown to directly bind them through its collectin binding domain28 and thereby contribute to their phagocytosis by lung-resident innate cells.28 This is important for clearance of pathogens including respiratory syncytial virus.29 Furthermore, SP-D can induce activation of lung DC, as well as promote inflammatory cytokine production and chemotaxis of macrophages.28 SP-D also exhibits some anti-inflammatory properties in some contexts including inhibition of smooth muscle cell cytokine production as well as preventing mast cell degranulation.28 SP-D can exist in both a monomeric and multimeric form and it remains unclear to what extent this contributes to its function in disease.28
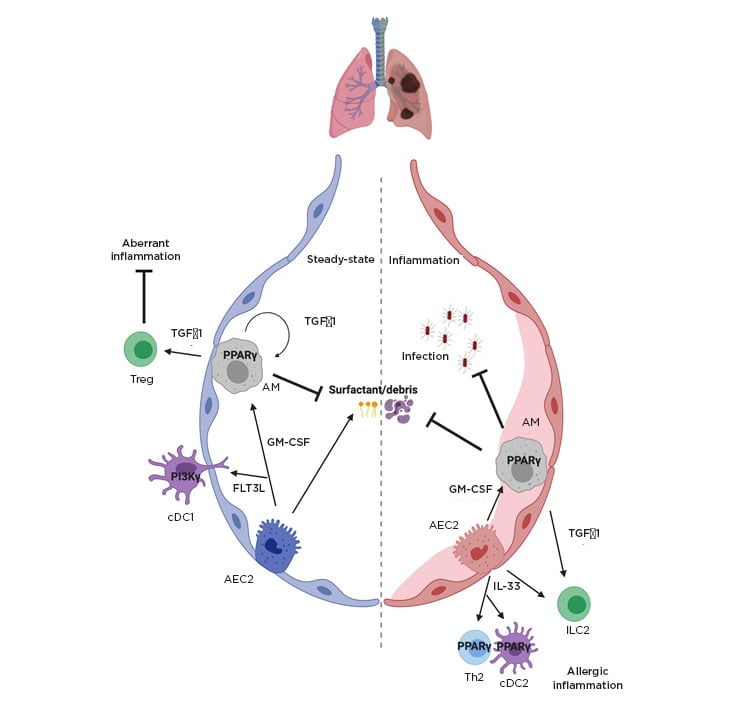
Figure 2: Key host regulators of lung-specific inflammation.
Key host factors controlling tissue-specific lung immunity in the steady-state and in inflammation are shown including transcription factor PPARγ, the cytokines IL-33, GM-CSF, and TGFβ1.
AEC2: type 2 alveolar epithelial cell; AM: alveolar macrophage; cDC1/2: conventional dendritic cell 1/2; FLT3L: FMS-like tyrosine kinase 3 ligand; GM-CSF: granulocyte-macrophage colony-stimulating factor; HIF1/2α: hypoxia-inducible factor 1/2α; ILC2: Type 2 innate lymphoid cells; PPARγ: peroxisome-proliferator activated receptor; TGFβ: transforming growth factor β; Th: T helper; Treg: regulatory T cell.
Similar to SP-D, SP-A has also been shown to directly opsonise respiratory bacteria and viruses and thereby contribute to pathogen clearance.30 However, it seems to have significant immunoregulatory properties because of its capacity to directly bind IFNγ and thereby suppress Type 1 immune responses.30 Furthermore, SP-A can also suppress lung DC activation and maturation in different contexts.30 While the role of the surfactant proteins in respiratory disease is increasingly studied, the importance of individual surfactant lipids in modulating the immune system remains largely unclear. More research will be necessary to investigate changing lipid landscapes in chronic lung diseases such as idiopathic pulmonary fibrosis and how this controls aberrant inflammation.
Transcription Factors: Peroxisome Proliferator-Activated Receptor γ
As a result of the lipid-rich environment, especially in the alveolar space, as described above, several host factors which are regulated by lipid ligands have been identified to play an important role in regulating lung immunity.
A key example in this context is peroxisome-proliferator activated receptor γ (PPARγ). This lipid-activated transcription factor is classically associated with regulating adipogenesis; however, in recent years multiple distinct functions of PPARγ have emerged in the lung.31 PPARγ can be highly expressed by several cell types in the lung including AM,32 DC,33 epithelial cells,34 as well as T cells.33,35
In nonhaematopoietic cells, a mechanistic understanding of PPARγ is still largely lacking but there is some evidence for a role in the development of emphysema34 and a minor role in lipid metabolism of club cells.36By contrast, in AM it was shown to be crucial for their development by regulating differentiation of lung fetal monocytes after birth.32 Perinatal induction of granulocyte-macrophage colony-stimulating factor (GM-CSF) expression in lung epithelial cells in turn induces PPARγ in lung fetal monocytes, which then give rise to a self-renewing AM compartment.32 Because of the essential role of AM in the maintenance of gas exchange during infection,37 PPARγ-deficiency leads to a highly increased susceptibility to influenza virus infection despite intact antiviral immunity37 which was associated with increased inflammation38 as well as impaired resolution of inflammation after infection.39 Apart from AM, PPARγ was also shown to play a key role in asthma where it is required for the induction and exacerbation of allergic lung inflammation.33 Contrary to early indications that PPARγ agonists dampen allergic immunity,40 PPARγ was shown to regulate the capacity of lung DC to trigger T helper (Th) 2-driven inflammatory responses, including eosinophilia and mucus production.33 It was also found to promote IL-33-induced effector function of Th2 cells themselves,33,35 suggesting a broader role in the development of allergic disease in the lung. Indeed, in human T cells PPARγ was shown to drive a specific Th2 effector programme characterised by high expression levels of IL-9, which in turn strongly correlates with allergic inflammation in the skin.41 While the inducers of PPARγ expression have been increasingly identified, the lipid ligands driving transcription factor activity in vivo remain largely unknown and more research is warranted to define these regulators in lung immunity. However, there is increasing evidence linking PPARγ to the HIF family of transcription factors,42suggesting a potentially indirect regulation of PPARγ activity by oxygen, but this is yet to be addressed in a mechanistic manner.
Tissue-Specific Cytokines: GM-CSF, IL-33, and TGFβ1
GM-CSF has recently emerged as a key orchestrator of lung immunity both in homeostasis as well as in inflammation. In the steady state it is mainly expressed by Type II alveolar epithelial cells,43 and is required for the development of AM37,44,45 and DC46,47 as well as to regulate the pool size of AM.48 Indeed, congenital deficiency of GM-CSF or its receptor leads to pulmonary alveolar proteinosis as a result of the absence of AM-mediated surfactant clearance.49 In inflammation, GM-CSF has shown to be crucial for maintenance of gas exchange during pulmonary viral infection37 and promote survival against pulmonary bacterial pathogens.50,51 Indeed, inhalation of GM-CSF is being explored as a potential treatment for coronavirus disease (COVID-19) in the hope of improving lung function. Furthermore, GM-CSF was shown to promote recruitment of granulocytes to the lung in allergic asthma.47 Interestingly, the protective function of GM-CSF in bacterial infection seems to depend on the presence of the microbiota through nucleotide-binding oligomerisation domain-containing protein 2-mediated stimulation of AM.50 This highlights the importance of key environmental factors for regulating lung immunity.
Another key element regulating the lung microenvironment is IL-33. While generally thought of as an alarmin and indicator of tissue damage, in the lung it is already highly expressed in the steady-state by lung epithelial cells and is critically important in the induction33 and exacerbation of allergic immune responses, including the development of asthma.52 Indeed, IL-33 single nucleotide polymorphisms are strongly associated with asthma in humans.53 In this context, IL-33 appears to be of particular importance in early life54 and involved in promoting virus-induced asthma exacerbations.55 Furthermore, IL-33 is important in promoting ILC2-mediated tissue repair after infection56 and regulatory T cell-mediated tissue homeostasis after lung injury.57It is also associated with disease severity in COPD58 and promoting detrimental inflammation in lung fibrosis59 via its effect on ILC2 and lung macrophages.60 Overall, through its strong role in promoting lung tissue-specific Type 2 immune responses it can thus be beneficial or harmful, depending on the disease context.
Furthermore, another key cytokine controlling tissue-specific immune responses in the lung is TGFβ1; an important regulator of lung development and in respiratory disease is classically associated with driving epithelial to mesenchymal transition in idiopathic pulmonary fibrosis.61 Recently, however, additional important roles in regulating lung immunity in health and disease have emerged. TGFβ1 is a key driver of AM differentiation and TGFβ receptor signalling, inducing PPARγ expression together with GM-CSF which then leads to full maturation of AM.62 AM themselves express TGFβ1 in the steady state, which is important for induction of allergic airway inflammation-suppressing regulatory T cells.63 Conversely, TGFβ1 promotes ILC2-mediated induction of airway hyperresponsiveness64 in asthma as well as IL-9 producing Th cells which drive lung structural remodelling in this context.65 In pulmonary viral infection, TGFβ1 has a detrimental role attributable to its suppression of early antiviral immunity;66 however, it also limits immunopathology later on and thus contributes to improved survival.67 In patients with acute respiratory distress syndrome, TGFβ1 in the bronchoalveolar lavage serves as a marker predicting poorer survival, but the underlying mechanisms remain unclear.68 Because of the complexity and the pleiotropic nature of the biology of TGFβ1, more research is necessary to understand its cell type and context-specific roles. In particular, the role of TGFβ1 in other common lung disorders, such as COPD, needs to be addressed on a more mechanistic level.
Lung Specific Signalling Factors: PI3Kγ
There is emerging evidence that various tissue cells of similar lineages use different signalling modalities to respond to key growth factors or hormones. This level of complexity in the signalling cascade downstream of common growth factor receptors is exemplified in the lung by the role of phosphoinositide receptor γ in controlling lung-specific development of DC.69 While PI3K signalling in general is known to regulate a wide myriad of processes, there is accumulating evidence that the different types of PI3K proteins have distinct and overlapping roles in different cell types and different microenvironmental niches.70 In the lung, DC as key innate sentinels of the immune system strictly require intact PI3Kγ to respond to the FMS-like tyrosine kinase 3 ligand,69 a key modulator of haematopoietic immune development.71 Deficiency in PI3Kγ leads to strongly impaired development of lung DC,69 which translates into increased susceptibility to viral infection because of impaired induction of antiviral immunity.72 PI3Kγ is not required for Fl3L-induced differentiation in closely related DC populations in other organs such as the skin or the intestine.69 This suggests that lung-resident cells harbour unique signalling networks that distinguish them from cells in other tissues. While the microenvironmental factor that drives PI3Kγ expression in lung DC is still unclear, it suggests that understanding tissue-specific signalling for each given cell type is crucial and molecular mechanisms in one tissue cannot be easily translated to another organ system. This emphasises that more research is necessary to identify lung-specific signalling regulators in other key cell lung-resident immune cell types.
Lung Structure and Tissue Organisation
Another key element that determines the uniqueness of the lung immune system is the need to adapt a highly structured environment with large airways, bronchi, bronchioles, and alveoli. While the understanding of how localisation within a tissue determines immune function is still in its infancy, there is emerging evidence that in the lung the function of immune cells is dependent on their localisation. For example, antigen uptake by lung DC is primarily restricted to cells localised close to alveoli, while subsequent inflammation develops more in the vicinity of the airways as a result of preferential recruitment of additional antigen-presenting cells and T cells in the context of allergic airway inflammation.73 Indeed, it appears that the proximity to the airways directly impacts DC behaviour in vivo.74 Adding further complexity to understanding the importance of local tissue organisation are changes to the tissue architecture during inflammation, such as airway remodelling in asthma or the emergence of tertiary lymphoid organs in lung infection. Understanding how the local microenvironment impacts lung immunity in health and disease is a key area of future research.
FUTURE PERSPECTIVES
Having described what unique environmental and intrinsic features regulate the respiratory immune system, the logical next step is to identify what the implications are for the future, from a research and therapeutic perspective. To understand the overall complexity of respiratory immunity in health and disease, more systematic approaches and methodologies need to be employed and with the advent of omics technologies this is becoming increasingly feasible. Comprehensive characterisation of healthy and disease states using single cell RNA sequencing will provide a more comprehensive picture of cellular composition, cell states, and cell–cell interaction networks. Especially emerging methods that incorporate spatial features will pave the way for a more complete understanding of how lung immunity contributes to disease. Such comprehensive analyses of different organ systems will also allow a more global view of what unique factors control respiratory immune responses as opposed to other tissues. This is of importance as it will facilitate the development of targeted therapies against pulmonary diseases. Apart from using monoclonal antibodies to inhibit host-intrinsic factors, which promote respiratory pathologies, the emergence of using other approaches that modulate lung immunity in a specific manner will be crucial for the development of better therapeutic measures. Indeed, understanding how the lung immune system interacts with other complex networks such as the respiratory microbiota will be crucial to understand the complex aetiology of some lung diseases such as asthma, COPD, or fibrosis. Many novel therapeutic approaches will include manipulating the respiratory microbiota using probiotics as well as microbiota-derived small molecule metabolites. While a mechanistic understanding of the role of the respiratory microbiota is still lacking, with emerging evidence for its importance in lung disease, it will be likely of great benefit to manipulate its composition and function to achieve a more healthy configuration and prevent disease-associated dysbiosis.






