
Symposium Summary
Viral lower respiratory tract infections (LRTI) are a leading cause of hospitalizations and death worldwide, placing significant pressure on healthcare systems. In this article, two pulmonary critical care specialists explored the prevalence and global burden of viral LRTIs, focusing on recent advancements in understanding the role of dysregulated host immune response and hyper-inflammation. Specifically, they focused on the relevance of alarmin cytokines and their association with adverse patient outcomes.
This article reviews the presentations delivered during an industry theater session that took place at the American Thoracic Society (ATS) 2023 International Conference, held in person in Washington, D.C., USA, in May 2023. The primary objective of this session was to provide a comprehensive overview of the impact and burden imposed by severe viral LRTIs. Pratik Sinha, an Assistant Professor in Anesthesiology at Washington University in St. Louis, Missouri, USA, provided valuable insights into the drivers behind the progression of viral LRTIs, which result in significant mortality and morbidity, imposing a considerable clinical burden with unmet needs. Nuala Meyer, an Associate Professor of Medicine at the Hospital of the University of Pennsylvania, Philadelphia, USA, shed light on the dysregulated host immune response and hyper-inflammation triggered by viral infections, which serve as key drivers of disease progression and severe patient outcomes. Sinha also discussed the potential role of alarmin cytokines, particularly IL-33, in influencing the severity and progression of respiratory viral infection. These cytokines may also serve as pleiotropic regulators of the host immune response to viral LRTIs.
Severe Viral Lower Respiratory Tract Infections Pose a Significant Burden on Patients and Healthcare Systems
Pratik Sinha
Severe viral LRTIs pose a substantial global burden, resulting in hospitalizations and death.1-5 This burden is particularly evident in the winter months, when seasonal RTIs caused by multiple viruses such as influenza, COVID-19, respiratory syncytial virus (RSV), metapneumovirus, and parainfluenza become prevalent (Figure 1).6 These infections continuously challenge the resilience of healthcare systems worldwide, leading to a surge in emergency hospitalizations and intensive care unit (ICU) admissions.6-8
Sinha described the impact of influenza in the USA prior to the COVID-19 pandemic. In 2019–2020 there were an estimated 36 million cases of influenza, with 25,000 deaths.1 Despite the surge in COVID-19 cases during the pandemic in 2020–2021, with around 147 million cases and 921,000 deaths,2 the burden of influenza has continued to persist. Preliminary estimates for 2022–2023 are up to 50 million cases of influenza and up to 56,000 deaths.3 According to Sinha, these numbers resonate with healthcare practitioners working in ICUs across the USA, highlighting the ongoing challenges posed by viral LRTIs.
The concern regarding viral LRTIs extends beyond mortality and encompasses substantial hospital admissions. Sinha emphasized that considering future burden, it is crucial to recognize that it is “not just influenza that we should be thinking about.” In the USA, it is projected that up to 630,000 patients may require hospitalization for influenza-associated LRTIs in 2022–2023,3 along with approximately 7.5 million cases of COVID-19.2 Additionally, for respiratory syncytial virus (RSV), the estimated annual number of patients hospitalized is up to 80,000 in those <5 years old and as many as 160,000 in those aged ≥65 years.9
Despite the recurrent peak periods in multiple viral pathogens, including influenza, severe acute respiratory syndrome coronavirus 2, and RSV, leading to RTIs (Figure 1),6 the timing and intensity of respiratory viral activity can vary and is not precisely determined from one season to the next.10-12 Sinha noted that this variability makes it “tricky to predict when in the winter these peaks will occur, and how large these peaks are going to be,” thus making optimal and efficient resource allocation of hospitals difficult, including staffing,4 and contributing to the financial burden.13 Therefore, the prevention and effective management of seasonal LRTIs is fundamental to reducing the demand on healthcare services.4
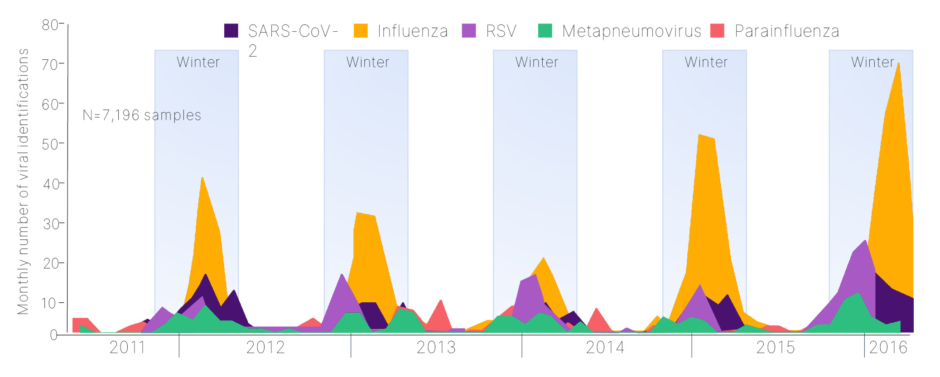
Figure 1: Seasonality of lower respiratory tract infections.6
Adapted from Visseaux et al.6
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
Sinha also emphasized the importance of considering the burden on patients. A study presented at the ATS 2023 International Conference, which summarized real-world evidence from a retrospective analysis of a USA claims database, assessed ICU admissions and outcomes associated with viral LRTI (influenza and COVID-19) before (2015–2019; n=49,317) and during (2020–2021; n=236,661) the COVID-19 pandemic.14 The study showed an increased burden on an already overwhelmed healthcare system in light of the COVID-19 pandemic. It demonstrated a rise in ICU admissions, escalating from 37% pre-pandemic to 44% during the pandemic. Additionally, there has been an increase in the utilization of invasive mechanical ventilation and extracorporeal membrane oxygenation, from 12% pre-pandemic to 25% during the pandemic, and a heightened mortality, escalating from 8% pre-pandemic to 22% during the pandemic.14 Influenza accounted for the majority of pre-pandemic cases, while COVID-19 contributed to the substantial increase in cases during the pandemic, amplifying the burden.14 Sinha highlighted the need to understand the impact of COVID-19 on seasonal pressures in conjunction with other RTIs in the future.
Sinha also noted that viral infections are known to “smoke out” non-respiratory symptoms such as palpitations, chest pain, dyspnea on exertion, and exercise intolerance.15 Concurrent infection with influenza has been associated with acute cerebrovascular events, such as ischemic stroke,16 as well as cardiovascular events, including myocarditis, acute myocardial infarction, heart failure, sudden cardiac arrest, and acute coronary syndrome.15,17-19
The Concept of Heterogeneity in Viral Lower Respiratory Tract Infections
There is a large degree of heterogeneity in response to viral LRTIs among patients, indicating that not every patient with an infection experiences the same outcomes, with marked differences in length of stay, O2 support, and other hospital trajectories.20 Thus, it is important to note that not all viral infections should be managed the same way.20 This heterogeneity may arise from multiple pathogens or host-driven responses, resulting in differing disease states, and can lead to severe complications, such as acute respiratory distress syndrome and death.20 For example, Sinha stated the need for respiratory support can differ substantially among hospitalized patients over time for those with COVID-19 pneumonia compared with influenza pneumonia.20 Sinha also stated that the best approach is to reduce the need for hospitalization.
Better understanding the dysregulated host immune response to viral LRTIs may present opportunities to improve patient outcomes.21
Treatment options for viral LRTIs are limited, and the unmet patient need is substantial. Sinha noted the need for more treatments and infection prevention programs. The COVID-19 pandemic has highlighted the potential benefits of targeting host-response inflammation.22 Sinha summarized that selecting the right patients at the right time with the right therapeutic approach is crucial.
Dysregulated Host Immune Response is the Driver of Disease Progression and Severe Patient Outcomes
Nuala Meyer
Appropriate host defense against viral respiratory infections is a delicate balancing act. Dysregulation of the host immune response contributes to progression and severity of the disease.23,24 Meyer discussed the intricacies and complexities associated with a dysregulated host immune response in viral LRTI, explaining that viral infections occur along a spectrum. During the early infection phase, most individuals are asymptomatic or experience mild symptoms such as fever, cough, myalgia, fatigue, sore throat, and headaches, which may persist for several days.23 However, clinicians encounter patients that develop infections in the lower airways, resulting in more severe symptoms of dyspnea, hypoxemia and tachypnea, which may result in pneumonia and respiratory failure necessitating hospitalization.23 Progression of the disease is driven by dysregulated host immune response to the infection and a dysregulated local and systemic inflammation. This phase, as described by Meyer, is also defined as sepsis, and leads to lung tissue damage, respiratory failure, acute respiratory distress syndrome, shock, and multi-organ failure, often resulting in death.23,24
Meyer emphasized the importance of considering how viral infections enter the human body and which host tissues are targeted, as well as the inflammatory pathways that are triggered.24 Respiratory viruses primarily target various lung epithelial cells, alveolar cells, endothelial cells, and immune cells.24 The initial point of contact for these viruses is typically the nasal mucosa and respiratory epithelium, as the virus travels down the nasal passage. These tissues are constantly “exposed to the outside world, always surveying,” and play a vigilant role in viral detection, triggering numerous inflammatory pathways (Figure 2).24,25
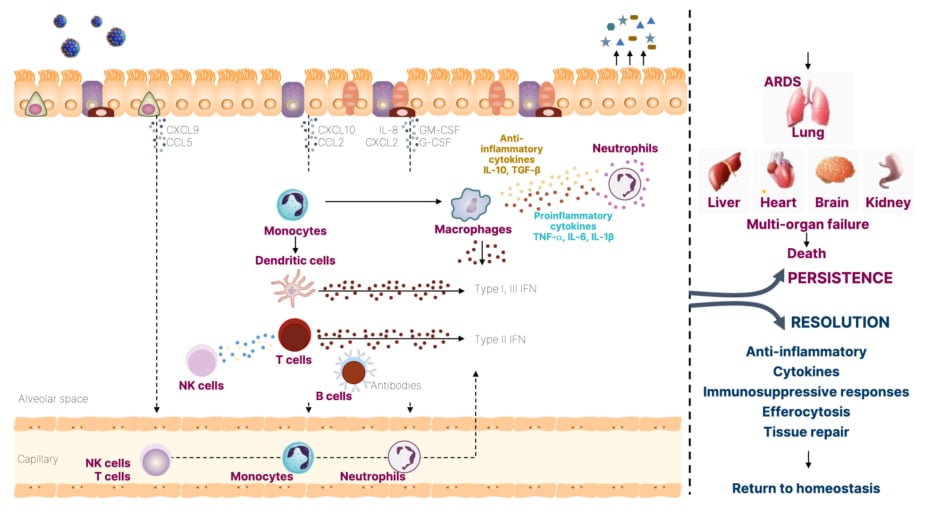
Figure 2: The inflammatory signaling pathways in lung epithelium and immunopathology during viral infections.24,25
Adapted from Clementi et al.,24 and Yang et al.25
ARDS: acute respiratory distress syndrome; CCL: chemokine (C-C motif) ligand; CXCL: chemokine (C-X-C motif) ligand; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon; NK: natural killer; TGF-β: transforming growth factor β.
The epithelium acts as a physical barrier and mounts protective secretory responses against viral infections, including the secretion of interferons (Type I-III) and pro-inflammatory cytokines such as IL-6, TNF-α, and chemokines.24 These cells recruit local innate immune cells such as neutrophils, monocytes, and dendritic cells, which activate the adaptive immune response involving natural killer cells, cytotoxic T cells, and B cells, inducing interferon (IFN)-γ secretion and antibody production.24 However, there can be a dysregulated immune response that can persist in some patients, leading to multi-organ failure.25 Meyer stated that with LRTIs, “the lungs are the most commonly affected organ,” but also provided examples of fatal influenza and RSV cases, where patients developed multi-organ failure along with circulatory dysfunction.23,25
In the context of the lung, inflammatory injury acts as a secondary indirect inducer of tissue damage following viral infection.26 Meyer emphasized the intricate nature of inflammation, highlighting the dysregulation that can occur in both the epithelium and endothelium. Inflammatory-driven injury increases the permeability of the epithelial barrier, allowing protein-rich edematous fluid to enter the alveolus.26 Meyer suggested that this process may “serve some function in pathogen-host defense by recruiting inflammatory cells to the alveoli.”26 However, this response can become dysregulated, leading to alveolar edema, respiratory failure, hypoxemia, and vascular injury, resulting in hypercapnia.26 These effects further impair the clearance of alveolar edema and can contribute to the development of acute respiratory distress syndrome.26
Evolutionary Paradigm: Defining, Describing, and Measuring Immune Dysregulation
Meyer emphasized the concept of innate immune dysregulation, which was traditionally viewed as a sequential process involving an initial hyper-inflammatory response characterized by excessive inflammation and elevated plasma cytokine levels, followed by a compensatory anti-inflammatory response.27 This hyper-inflammation can cause tissue damage, while dysregulated adaptive immune suppression may place patients at risk of secondary infections.27 Meyer clarified that these processes actually occur simultaneously within the same patient, and often those who exhibit “the most hyper-inflammatory response also display the most adaptive immune dysregulation.” Thus, immune dysregulation manifests as a sustained inflammatory state driven by dysfunctional innate immunity and suppressed adaptive immunity, which may contribute to mortality in the long term.27
Meyer highlighted the need to establish markers that can aid in identifying excess inflammation and dysregulated adaptive immunity. Potential candidate markers include elevated inflammatory cytokines,28 vascular activation,28,29 activated neutrophils,28 low reduced human leukocyte antigen monocytes,27,29 endotoxin tolerance,27 and apoptosis.28 These markers may indicate a dysregulated host response, yet Meyer emphasized that further research is required to identify the key markers, due to patient heterogeneity. Despite this heterogeneity, there are examples of consistent patterns observed across different pathogens. Translational studies have demonstrated that elevated levels of IL-6, for instance, show strong associations with disease severity and survival in patients with LRTIs. A single-center study of patients hospitalized with COVID-19 (N=1,484) in the USA, demonstrated increased IL-6 levels in severe cases and independently predicted survival.30 Similarly, elevated IL-6 levels were associated with disease severity in a cohort of patients hospitalized with influenza A virus subtype H1N1 infection (N=32), particularly those with acute respiratory distress syndrome (n=21).31 In children ≤5 years old who attended the emergency department (between September 1998–May 2008) with RSV infection (N=851), elevated IL-6 levels were also associated with disease severity.32 Similar results were observed for other pro-inflammatory cytokines, including IL-1β, IL-8, and TNF-α.30-32
Heterogeneity Among Severe Patients Hospitalized with COVID-19
However, heterogeneity in the host response can exist even within the same pathogen. As an example, a study profiling 125 patients hospitalized with COVID-19 compared them with healthy donors or individuals who experienced mild COVID-19 and recovered without requiring hospitalization.33 Using a uniform manifold approximation approach, approximately 200 immune features were distilled into two dimensions, which identified three immunotypes of COVID-19 associated with clinical outcomes.33 The findings demonstrated that patients who were severely ill clustered separately from healthy donors and those who recovered without hospitilization.33 Among the patients with severe illness, the distribution demonstrated more heterogeneity while identifying multiple clinical features.33 Those identified as ‘immunotype 1’, associated with higher severity of illness, organ failure (as measured by Acute Physiology, Age, Chronic Health Evaluation [APACHE] III scoring), and higher mortality, and corresponded with hyperactivated T cells, higher plasmablasts, and lower altered follicular helper T cells.33 ‘Immunotype 2’ represented “a more balanced immune response,” stated Meyer, with the emergence of memory B and T cells, while ‘immunotype 3’ showed little evidence of active B or T cell response, indicating the complex heterogeneity of the immune response with COVID-19.33
Additionally, heterogeneity has also been observed in the secreted plasma protein response to COVID-19, where a protein signature consisting of 14 plasma proteins strongly associated with severe disease (defined as illness that resulted in death, the need for high-flow nasal cannula, non-invasive or invasive positive pressure ventilation, or extracorporeal membrane oxygenation).34 Differential upregulation of protein expression (n=145), including tenascin-C, C-C motif chemokine ligand 7, transforming growth factor-α, and IL1-receptor like-1, and downregulation (n=49) of IL12-β, TNF superfamily 11, and matrix metallopeptidase 9, were observed after correcting for multiple comparisons.34 Meyer identified that these proteins could be integrated into a “protein severity score” that could provide information about a patient’s prognosis. This proteomic score was used to evaluate heterogenous treatment effect by predict disease severity or death in an observational study involving patients hospitalized with COVID from Massachusetts General Hospital, Boston, USA.34 The data demonstrated an effective modification in the relationship between the protein score, stratified by corticosteroid receipt, and the prediction of severe disease or mortality at 90 days, with an area under the receiver operating characteristic of 0.88 (95% confidence interval [CI]: 0.83–0.93).34 This was validated in an external dataset, yielding an area under the receiver operating characteristic of 0.92 (95% CI: 0.87–0.95).34 Of note, patients with a low predicted severity protein score who received corticosteroids were associated with higher mortality, suggesting a disruption of the regulated host response to the virus.34
Meyer summarized that in order to make progress, key features of the viral disease need to be identified, and targeted precision medicine may be used for the treatment of patients with severe LRTIs. Similar achievements have been made in other complex traits, where the identification of biomarkers has driven substantial improvements in patient care, management, and outcomes.35-37 To facilitate this progress, Meyer identified the three major advances are needed: a better understanding of the drivers of severe outcomes and their causality;35 quantitative traits that can be measured to indicate patients with significant of dysregulated inflammation; and the ability to promptly sort or classify patients to identify which therapy is suitable for each individual.
The Role of the Alarmin IL-33 in Viral Lower Respiratory Tract Infections
Pratik Sinha
Alarmins are a group of proteins and peptides released upon cellular injury.38-40 They are effector molecules that trigger downstream activation of inflammation and act as a chemotactic agent, stimulating both the innate and adaptive immune system.38 Alarmins include thymic stromal lymphopoietin, IL-25, and IL-33, which are central regulators of the immune response.38
Sinha focused specifically on the role of IL-33, noting that it is part of the IL-1 superfamily and acts as a danger-associated molecular pattern protein.40,41
Following damage to the airway epithelium induced by several triggers, including viruses, toxins, allergens, or smoking,41 there is a rapid release of bioactive IL-33 into the extracellular milieu as cells die.42 Sinha stated that IL-33 acts via two distinct receptor pathways (Figure 3).43
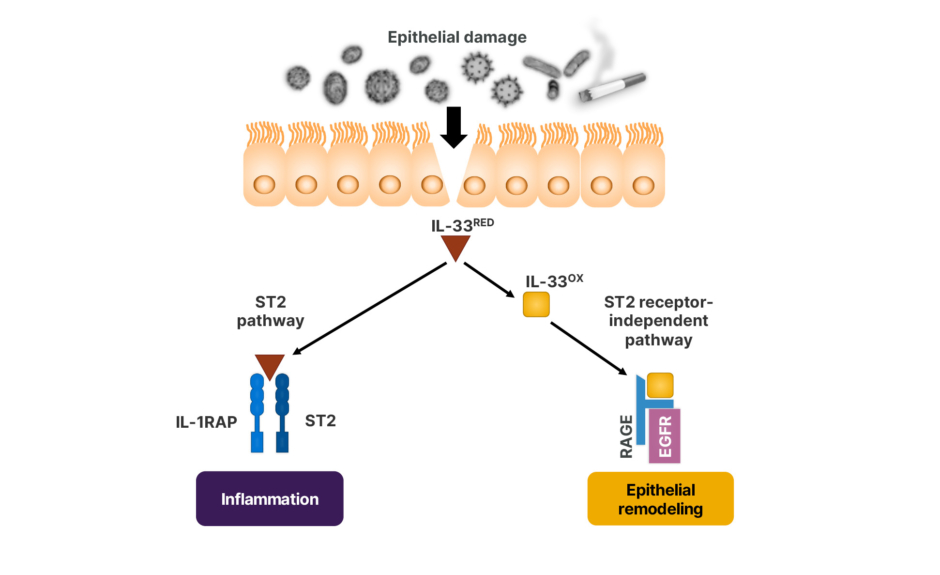
Figure 3: The distinct receptor pathways of IL-33.41-43
EGFR: epidermal growth factor receptor; IL-1RAP: IL-1 receptor accessory protein; IL-33OX: oxidized IL-33; IL-33RED: reduced IL-33; RAGE: receptor for advanced glycation end products; ST2: serum stimulated-2.
In the “canonical pathway,” reduced IL-33 binds to ubiquitous serum stimulated-2 (ST2) receptors, forming a ternary IL-33/ST2/IL-1 receptor accessory protein complex that induces a broad downstream inflammatory cascade.41,42 Under the “non-canonical pathway,” oxidized IL-33 is unable to bind to ST2.42 Instead, it binds to the receptor for advanced glycation end products and epidermal growth factor receptor complex on epithelial cells, leading to epithelial repair, regeneration, and remodeling.43 Sinha added that IL-33 is constitutively expressed in airway epithelial and endothelial cells, indicating a homeostatic role in barrier repair and maintenance. However further research is required to establish IL-33’s potential as a biomarker in lung epithelial injury.38,44-46 In addition, IL-33 is a pleiotropic amplifier of inflammation and may be an upstream regulator of host damage, leading to epithelial injury.44,45 However, Sinha pointed out that “a better understanding of the difference between a regulated versus dysregulated response is
still required.”
Upon rapid release from damaged cells, the reduced IL-33 binds and signals through the ST2 receptor expressed by both innate and adaptive immune cells.45 By binding to ST2, IL-33 activates these immune cells to produce multiple cytokines and growth factors that promote and regulate both local and systemic immunity.45 Additionally, IL-33 expression is known to be upregulated in viral respiratory infection, and various inflammatory diseases such as asthma, where it increases airway hyperresponsiveness.44,45 Furthermore, Sinha added that IL-33 plays a role in the interaction with fibroblasts during epithelial airway regeneration.45,45
Sinha stated that IL-33 is “a molecule that is effective in homeostasis, but is involved in triggering the immune system upon damage.” Sinha also mentioned that the function of IL-33 is “related to the timing and conditions under which it is found communicating with other cells.” In addition, Sinha summarized that the “activation of the IL-33 pathway may be implicated in the dysregulated inflammation related to airway tissue damage, as seen in viral infections.”
There is evidence supporting the pleiotropic effects of IL-33. In a study of natural killer T cells co-cultured with IL-33, it was demonstrated that downstream protein biomarker response of cytokines IL-4, IL13, IL5, IL2, IFN-γ, and TNF-α increased in a dose-dependent manner, demonstrating that IL-33 enhances the effector function of human invariant natural killer T cells.47
One hypothesis of the IL-33-mediated mechanism of injury, suggests that epithelial injury caused by a virus may lead to the rapid release of reduced IL-33, which may drive dysregulated inflammation and epithelial and endothelial dysfunction.43,46 This, in turn, has a downstream impact on ST2 receptors, resulting in an increase in inflammatory cytokines that further signal epithelial injury (Figure 4).43,46
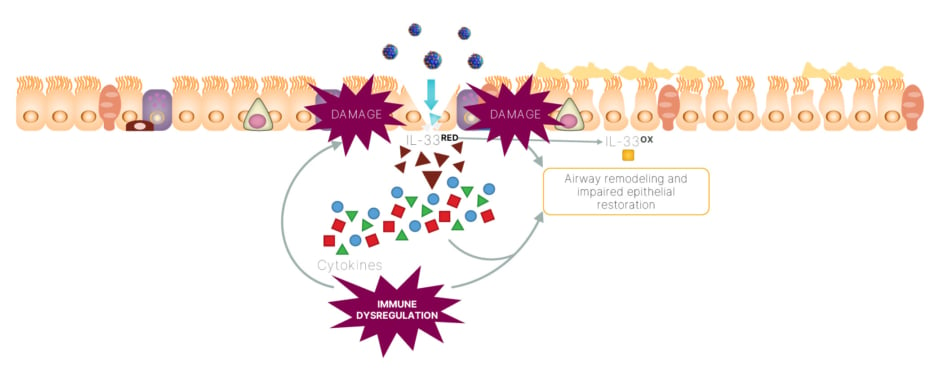
Figure 4: An IL-33 feedforward loop may drive dysregulated inflammation, as well as epithelial and
endothelial dysfunction in viral infections.43,46
IL-33OX: oxidized IL-33; IL-33RED: reduced IL-33.
Sinha reviewed some evidence suggesting a potential role for IL-33 in viral LRTIs. In a study of human primary bronchial epithelial cells, it was determined that IL-33 release was induced by viral infections (influenza A, RSV, and rhinovirus) and this induction increased in a time-dependent manner (1–72 hours), highlighting a potential role of IL-33 across a broad range of viral RTIs.48 Additionally, in a study of patients hospitalized with COVID-19 (N=40), peripheral blood obtained during the 30 days post symptom onset showed that serum IL-33 levels correlated with disease severity, with higher levels in moderate/severe disease compared with mild disease (p=0.0211).49 Similar results were observed for IFN-α2, IL-6, and IL-10.49 There is also evidence of pathogen-dependent host responses. In a study comparing IL-33 levels in adults with influenza A (n=24) and influenza B (n=48), serum IL-33 levels were significantly higher in patients with influenza A (p<0.05).50
IL-33 is also associated with more severe disease in infants and children with RSV infection. A study including infants <5 years with LRTIs caused by RSV (n=29) showed significantly higher expression of IL-33 (p=0.029) in nasopharyngeal aspirates compared to those with upper RTIs (n=12).51 Similar findings were observed for nasopharyngeal IL-8 levels, but not for IL-3 or thymic stromal lymphopoietin levels.51 Similarly, another study found that IL-33 or ST2 levels in nasopharyngeal secretions were associated with the severity of acute LRTI in children <5 years (p<0.001).52 The study also showed a higher risk of mechanical ventilation with a risk ratio of 2.89 (95% CI: 1.83–4.57) for IL-33 and a risk ratio of 4.57 (95% CI: 1.78–11.70) for soluble suppression of tumorigenicity 2.52
Sinha summarized the need for further investigation to establish the mechanisms involved and determine whether IL-33 is involved in dysregulated inflammation and epithelial and endothelial dysfunction pathways. Additionally, there needs to be better understanding of the kinetics of IL-33, as well as better understanding in the molecular epidemiology of viral LRTI populations.
Summary
In conclusion, LRTIs are a leading cause of hospitalizations and death globally, placing a significant burden on healthcare systems.1–5 The dysregulation of the host immune response plays a critical role in the severity and progression of LRTIs.23,24 Importantly, IL-33 has emerged as a potential pleiotropic regulator of dysfunctional host viral response in patients with LRTIs.44
Compliance number: Z4-55877 / June 2023






