Meeting Summary
This symposium took place during the 2022 meeting of the European Respiratory Society (ERS). Focusing on new frontiers in chronic obstructive pulmonary disease (COPD), the speakers discussed the current understanding of the heterogeneity of COPD, the spectrum of inflammatory pathways in COPD pathophysiology, and the evolving clinical landscape in COPD. Alvar Agustí, Professor of Medicine at the University of Barcelona, Spain, introduced the concept of a gene, environment, and time (GETomic) perspective of COPD, ending with the trajectome, or range of lung function trajectories, through the lifespan. Stephanie Christenson, Assistant Professor of Pulmonology at the University of California, San Francisco, USA, followed by exploring the role of inflammation in COPD, highlighting that it is a heterogeneous disease with broad inflammatory pathways. This includes types 1, 17, and 2 inflammation; a wide range of immune effector cells beyond eosinophils; and key cytokines such as IL-33, IL-4, IL-13, and IL-5, which mediate the pathophysiology of COPD in response to cigarette smoke and other environmental insults. The need for continued investigation of molecular phenotypes was expanded on further by MeiLan Han, Professor of Medicine at the University of Michigan, Ann Arbor, USA, who explored the knowns and unknowns in the standard-of-care management of patients with COPD, highlighting the evolving understanding and identification of disease endotypes and phenotypes; poor understanding of COPD disease progression; the importance of smoking status in response to currently available treatments; and significant unmet need for additional treatment, even among patients on triple therapy. Dave Singh, Professor of Respiratory Pharmacology at the University of Manchester, UK, then discussed active areas of investigation in the development of additional treatments for patients with COPD.
A GETomics Perspective of Chronic Obstructive Pulmonary Disease: What Is It?
Alvar Agustí
All human diseases, including COPD, are the end result of gene–environment interactions.1 However, this conventional model is being modified. COVID-19 demonstrated that mortality is higher in older versus younger individuals. The viral and environmental exposures were the same, but the obvious difference was age. Thus, the additional factor of time was proposed as part of the GETomics approach.1
GETomics contextualises the complex interaction between genes (G) and the environment (E) over time (T).1 GETomics is a holistic strategy that integrates basic omics (genomics, epigenomics, proteomics, and metabolomics) with clinical omics (e.g., phenomics, physiomics, and radiomics), and the exposome over time.1 The time component is critical, because when a given gene–environment interaction occurs, combined with previous and cumulative gene–environment interactions over the lifespan of an individual, it can influence the final biological and clinical outcome.1
In the context of COPD, the end result of GETomics is the trajectome, defined as the spectrum of possible lung function trajectories across the lifespan, including supranormal, normal, pseudonormal, below normal, and early decline.1 Both abnormal lung development and an increased rate of lung function decline associated with ageing can coexist and result in COPD in adulthood.2 Individuals with lung function trajectories below the expected normal range due to suboptimal growth and development in childhood and adolescence, or due to early or accelerated lung function decline in adulthood, may have an increased risk of COPD.1
It is believed that if COPD is to be eradicated, a change that anticipates the diagnosis and treatment of the disease is needed.2 However, the current paradigm is that randomised controlled trials are conducted towards the tail end of the lung function trajectory. By conducting randomised controlled trials earlier in the COPD trajectome, the public health impact of the disease may be reduced by improving prevention, treatment, and prognosis at earlier time points.2
Advances in Our Understanding of Inflammation in Chronic Obstructive Pulmonary Disease
Stephanie Christenson
The pathology of COPD involves altered small (<2 mm) airways early in the course of disease.3 The classical understanding of COPD is that smoking, for example, results in injury that reprogrammes basal cells, causing small airway epithelium loss of secretory and ciliated cells and squamous and goblet cell metaplasia, contributing to altered innate immune defences, mucus secretion, and dysfunctional cilia.3 However, Christenson emphasised that processes leading to this pathophysiology are not always the same. More recent understanding of COPD reflects heterogeneity in how COPD may develop in different patients. Inflammation in COPD is a multifaceted process (Figure 1),4-26 involving goblet cell hyperplasia and mucus production, increased neutrophil levels, mast cell degranulation, and increased blood eosinophil levels, resulting in airway remodelling (e.g., fibrosis, airway smooth muscle contractility and thickening, and matrix metalloproteinase production).1-23 In addition, there are multiple inflammatory pathways in the lungs, with type 1/type 17 (neutrophilic) inflammation present in majority of patients and type 2 inflammation additionally present in 30–40% of patients with COPD.4-7,22,23
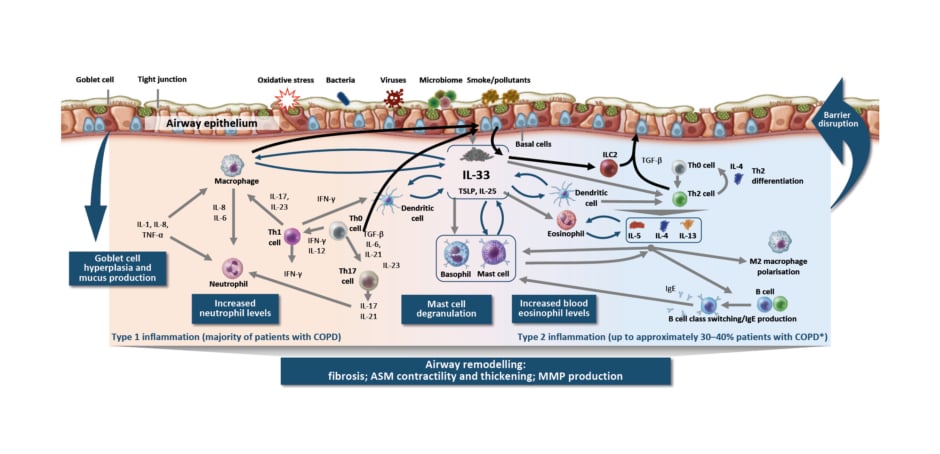
Figure 1: Inflammation in chronic obstructive pulmonary disease is a multifaceted process.4-26
*Based on sputum EOS ≥3% and blood EOS ≥2%.
ASM: airway smooth muscle; COPD: chronic obstructive pulmonary disease; EOS: eosinophil; IFN: interferon; ILC2: group 2 innate lymphoid cell; MMP: matrix metalloproteinase; TGF-β: transforming growth factor β; Th0: T helper type 0; Th1: T helper type 1; Th2: T helper type 2; Th17: T helper type 17; TSLP: thymic stromal lymphopoietin.
Type 1/type 17 inflammation in COPD is characterised by cells of the innate immune system that participate in the inflammatory response, including natural killer cells, dendritic cells, and the epithelial barrier.27 In particular, Christenson highlighted the increase in macrophage numbers in airway epithelium, tissue, bronchoalveolar lavage, and sputum.28 Macrophages also produce an elastolytic inflammatory response and increased levels of matrix metalloproteinases, resulting in damage to lung tissue.25,28 In addition, macrophages are unable to mount an adequate phagocytic response, which may result in impaired clearance of bacteria, in microbial dysbiosis, and, subsequently, in persistent inflammation.25 Similarly, neutrophil numbers are increased in the airway lumen, and in COPD, neutrophils exhibit increased migration and induce mucus hypersecretion.25,28
Christenson also highlighted the different molecular phenotypes in COPD and polarised adaptive immune responses. In stable obstructive disease, she noted that there are type 2-high, type 1-high, and type 17-high phenotypes. Type 2-high is what one would consider classically ’allergic’ steroid-responsive COPD, which is associated with eosinophilic inflammation.29,30 Type 1-high is classically ‘antiviral’, and is characterised by increased interferon, IL-2, and lymphotoxin-α, and by decreased lung function and increased bronchodilator response, independent of type 2 inflammation.31,32 Type 17-high is classically ‘antibacterial’ steroid-unresponsive COPD associated with neutrophilic inflammation.33 In IL-17-related inflammation in COPD, Christenson emphasised that neutrophilic inflammation, macrophage accumulation, and lymphoid neogenesis contribute to tissue destruction/emphysema, corticosteroid unresponsiveness, bronchial hyper-responsiveness, and lung function decline in COPD.34-36 These findings are based in part on research into the transcriptional response of the airway epithelium to IL-17. Christenson et al.33 generated a gene expression signature of IL-17A response in bronchial airway epithelial brushings from smokers with and without COPD, and related it to clinical and pathologic characteristics in two COPD studies: SPIROMICS and GLUCOLD. The IL-17 signature was moderately associated with sputum neutrophil counts in both randomised COPD trials.33 Additionally, in GLUCOLD, a higher baseline IL-17 signature was found to be associated with lack of forced expiratory volume in 1 second response to inhaled corticosteroid (ICS) over 30 months compared with a lower IL-17 signature.33 These results suggest that patients with COPD might be inflamed but not steroid-responsive, and represent a patient population that could be targeted via a different pathway.
Christenson also highlighted the role of alarmins, particularly IL-33, in COPD. Chronic cigarette smoke exposure has a pleiotropic effect and can stimulate an inflammatory response in COPD, resulting in increased release of IL-33 and altered expression of its receptor (ST2).37-39 IL-33 released from lung epithelial cells, fibroblasts, and blood vessel endothelial cells promotes immune response, airway hyper-responsiveness, and airway remodeling. Conversely, reduced IL-33 signalling by a rare loss-of-function variant protects against COPD.40 Additional evidence for IL-33 expression being important in COPD has been found in smokers; IL-33 expression in the lungs has been found to be highest in patients with severe COPD (Global Initiative for Chronic Obstructive Lung Disease [GOLD] IV).37 These findings provide the biological basis for the importance of IL-33 in the pathophysiology of COPD.
In comparison, type 2 inflammation in the airway in COPD is associated with basal cell, innate lymphoid cell, and Th2 cell feedback loops; activation of basophils, eosinophils, and mast cells; and inflammatory cell trafficking to tissues, including eosinophilic inflammation, mediated by IL-4, IL-13, and IL-5.4,7,8,10,14-18,20,21 Importantly, Christenson highlighted data on elevated eosinophils and their association with more frequent exacerbations in patients with COPD. Patients with elevated blood eosinophils (≥340 cells/µL, or 3.3%) had up to a three-times greater risk of exacerbations compared with patients without elevated blood eosinophils in a Copenhagen general population study.41 However, these data should be interpreted with caution because the proportion of patients with COPD with elevated blood or sputum eosinophils can vary, depending on various factors. Furthermore, evidence suggests IL-4 and IL-13 are key and central cytokines that can be elevated in stable COPD and during acute exacerbations.7,42-46 Elevated type 2 cytokines may result from the release of epithelial-derived cytokines thymic stromal lymphopoietin and IL-33 from airway epithelial cells, in response to cigarette smoke and/or viral infection.47 Christenson also highlighted the influence of the environment in the heterogeneity of exacerbation triggers, noting reduced COPD exacerbations observed with COVID-19.
In summary, Christenson highlighted that COPD is a chronic, progressive disease with a multifaceted inflammatory process encompassing non-type 2 and type 2 inflammation. Multiple key cytokines and a wide range of immune effector cells beyond eosinophils mediate the pathophysiology of COPD in response to cigarette smoke and other environmental insults. Furthermore, the heterogeneous nature of COPD highlights the need for continued investigation of molecular phenotypes, to facilitate a more targeted therapeutic approach.
Knowns and Unknowns in Chronic Obstructive Pulmonary Disease Management
MeiLan Han
Given the significant heterogeneity in COPD with respect to clinical presentation, physiology, response to therapy, decline in lung function, and survival,48 Han differentiated the roles of phenotypes, endotypes, and biomarkers in characterising different patient populations with COPD and how this may impact the management of COPD.
Han noted that in COPD, clinicians have focused on disease attributes such as symptoms, exacerbations, response to therapy, rate of disease progression, or death.48 In contrast, endotypes may identify a subtype of a condition, defined by a distinct functional or pathobiological mechanism. Han suggested that patients with a specific endotype would present themselves within phenotypic clusters of diseases.48,49 However, this has not been formalised in COPD.
The changing understanding of COPD brings a new way of characterising patients (Figure 2). Traditionally, patients with COPD were considered emphysemic ‘pink puffers’, noted for dyspnoea, air trapping, hyperinflation, and cachexia, whereas chronic bronchitic ‘blue bloaters’ were noted for chronic productive cough, respiratory infections, exacerbations, and being overweight.26,50,51 Current treatment-related phenotypes include the emphysema-predominant, frequent exacerbator, and bronchitis-predominant phenotypes.52,53 However, emerging endotypes and phenotypes are becoming increasingly recognised, such as neutrophilic, eosinophilic, and an additional emerging phenotype/endotype of former smokers.26,54 A better understanding of the inflammatory pathways involved, Han observed, may inform more precise treatment options.
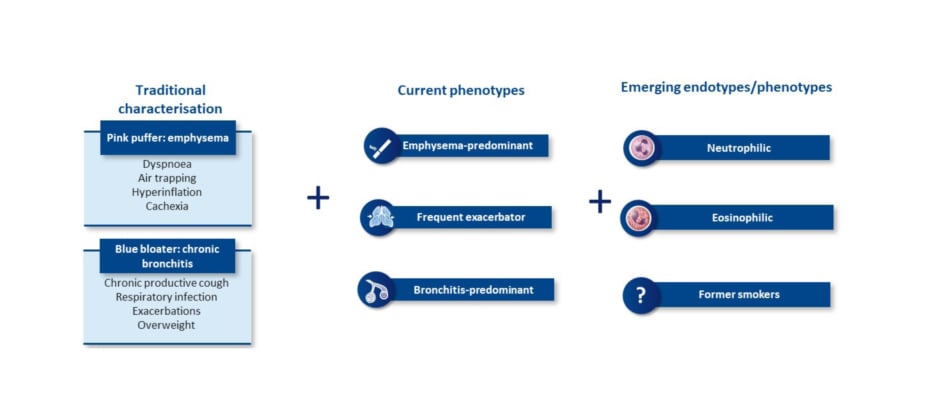
Figure 2: Chronic obstructive pulmonary disease is a complex and heterogeneous airway disease with multiple phenotypes and emerging endotypes.26,50-54
Elevated eosinophils are acknowledged as a marker of type 2 inflammation and an emerging endotype in COPD. However, Han cautioned that COPD and asthma can overlap, because type 2 inflammation occurs in both diseases. While patients with symptoms of asthma and COPD were previously diagnosed with asthma-COPD overlap, the latest guidelines (GOLD 2022, Global Initiative for Asthma [GINA] 2022, and Spanish COPD Guidelines [GesEPOC] 2021) emphasise that asthma and COPD are two separate diseases, and that some patients may present with characteristics of both.26,55-57
Having a biomarker would assist clinicians in understanding which patients may benefit from particular treatments. Evidence supporting the role of blood eosinophils as a potential biomarker to identify patients who may benefit from ICS were shown from two studies: an analysis of data from three randomised controlled trials of budesonide-formoterol in patients with COPD with a history of exacerbations and available blood eosinophil counts; and an analysis of the IMPACT trial, a Phase III, randomised, double-blind, parallel-group, multicentre study that modelled the relationship between blood eosinophil counts and ICS therapy on clinical outcomes in patients with symptomatic COPD.58,59 Across both studies, the magnitude of benefit or regimens on annual exacerbation rate increased in proportion to peripheral blood eosinophil count and by baseline blood eosinophil count quintiles with ICS-containing treatment, compared with non-ICS-containing treatment groups.58,59
Han cautioned that while the data for eosinophils are intriguing, the inflammatory characterisation of COPD based on biomarkers is not well established. GOLD recognises blood eosinophils as a biomarker for ICS use, but states that continuous cautious and realistic interpretation of the role of biomarkers in COPD is required due to the weak associations and lack of reproducibility in large cohorts.26 Blood eosinophils ≥300 cells/μL may help clinicians predict the likelihood of a beneficial response to addition of ICS for prevention of exacerbations, and blood eosinophils <100 cells/µL may identify patients with low likelihood of response.26 Despite their potential utility, blood eosinophils are not approved as a circulating biomarker. Indeed, fibrinogen is the only circulating biomarker approved to date by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), although they are mostly used by researchers to enrich patient populations.60 Therefore, despite recent interest, the concept of endotyping is still emerging in COPD, and there is a lack of established biomarkers to differentiate inflammatory profiles, further complicating the treatment decision-making process.
Han then highlighted how additional unmet needs and unknowns contribute to the complexity of managing COPD. For example, former smokers with COPD present a challenge because they still have accelerated lung function decline compared with never-smokers, and this decline has been observed for decades after cessation.61 Why this happens is not understood, but Han suggested that smoking may be a double-edged sword, where it causes damage in active smokers, but also an unexpected immunomodulatory effect that manifests when patients stop smoking. Patients may then be most vulnerable to symptoms and exacerbations immediately following smoking cessation.
Another area of unmet need is in the pharmacological management of patients with COPD, especially those with exacerbations who require follow-up management, as recommended in GOLD 2022.26 Recommendations for escalation include maximal or triple inhaled therapy, using peripheral blood eosinophil counts as a biomarker to guide ICS use.26
Many patients with COPD continue to experience exacerbations and symptoms despite maximal inhaled therapy, as evidenced by a post hoc analysis of the IMPACT study.62 Of patients on triple therapy, regardless of exacerbation history, 45–51% continued to experience moderate or severe exacerbations, and 55–61% did not achieve a clinically meaningful improvement in St. George’s Respiratory Questionnaire (SGRQ) total score, highlighting the need for alternative therapies for patients who continue to exacerbate or experience debilitating symptoms.62 In addition, there is also an unmet need for alternative therapies for patients with COPD who fail to meet local guideline criteria for triple therapy, and for those with blood eosinophils <100 cells/µL, and in whom addition of ICS is of limited benefit or contraindicated (Figure 3).26,63-65 In support of the previous observation of an emergent phenotype, former smokers have a unique treatment response, with the benefit of ICS and azithromycin more pronounced among former smokers, potentially highlighting an immunomodulatory effect that is offset by smoking.59,66
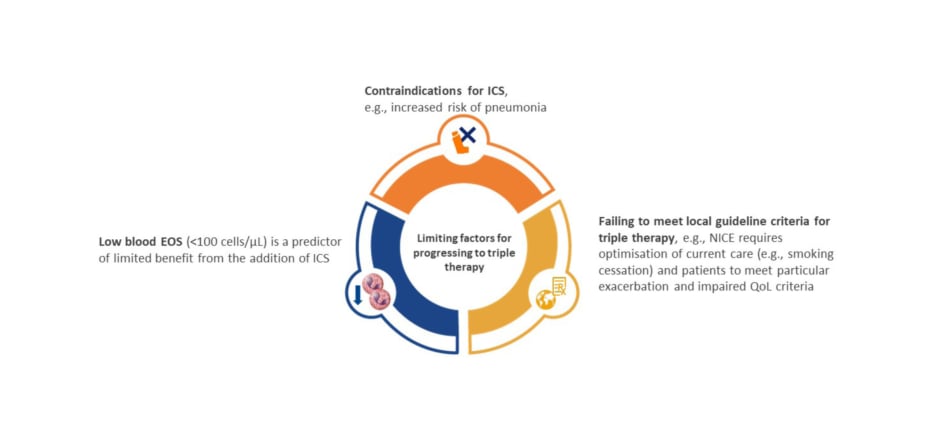
Figure 3: Unmet need: alternative therapies for patients for whom triple inhaled therapy is not advised, or is contraindicated.26,63-65
EOS: eosinophils; ICS: inhaled corticosteroid; NICE: National Institute for Health and Care Excellence; QoL: quality of life.
In summary, Han emphasised that understanding of disease endotypes and emerging phenotypes is evolving, as is the understanding of how to identify them. In patients with COPD, disease progression, independent of smoking status, is poorly understood. Smoking status not only impacts disease progression but may also affect treatment response to certain commonly used therapies. Current inhaled therapies reduce exacerbations and improve symptoms, but the need for additional treatment, even among patients on triple therapy, is significant. The potential role of molecules in clinical development for COPD is an active area of investigation.
Changing Treatment Paradigms: The Evolving Biologic Landscape in Chronic Obstructive Pulmonary Disease
Dave Singh
As noted earlier, a number of potential targets in COPD are being explored, with biologics against various inflammatory mediators being among the molecules in clinical development. While too extensive to discuss in detail, these clinical development programmes include different COPD patient populations.
Singh summarised ongoing investigations, recognising that previous clinical trials in COPD have suggested that blood eosinophil levels and smoking status may play a role in the magnitude of treatment response related to COPD exacerbations and lung function. There is an ongoing pipeline of emerging therapies targeting different aspects of the spectrum of inflammatory pathways involved in COPD, reflecting the heterogeneous nature of COPD. Singh shared his optimism for the future of COPD, highlighting that results from these COPD studies with different patient populations will positively impact understanding of disease pathophysiology, and facilitate individualised approaches to clinical management of COPD.
Agustí concluded the symposium by thanking his co-panelists and the audience.
MAT-GLB-2204689 V1
11/2022