![EMJ 5 [Supplement 1] 2020 Feature Image](https://www.emjreviews.com/wp-content/uploads/2020/12/EMJ-5-Supplement-1-2020-Feature-Image-940x563.jpg)
Meeting Summary
In this symposium at the virtual European Respiratory Society (ERS) International Congress 2020, co-chair Prof Sitbon introduced the symposium by highlighting that many patients with pulmonary arterial hypertension (PAH) are already classified as being in the World Health Organization (WHO) functional Class III/IV at diagnosis, as early symptoms are nonspecific. However, early diagnosis and management are important to improve prognosis. Furthermore, the importance of correctly diagnosing the aetiology of pulmonary hypertension (PH) was highlighted through two clinical cases from Dr Manes and Dr Blanco because the treatment options for these patients differ. This can be difficult, but Prof Gaine continued the symposium by presenting some pearls of wisdom to help with the differential diagnosis towards PAH. Prof Hoeper then said that risk assessment, at diagnosis and then 3–6 times monthly, is very important in PAH. Patients with low or intermediate risk of 1-year mortality should usually receive initial double oral combination therapy, as this has been shown to be superior to initial monotherapy, while high-risk patients should receive initial combination therapy including an intravenous prostacyclin analogue. Patients who are not low-risk at follow-up should have their therapy escalated (with an additional drug) to improve prognosis. Lastly, co-chair Prof Delcroix closed the meeting by reminding the audience of the importance of differential diagnosis, risk assessment, and treatment strategies.
Introduction
Professor Olivier Sitbon
The objectives of this symposium were firstly to recognise the difficult diagnostic journey faced by patients with PAH, to learn expert tips on how best to differentiate between the PH groups and ways to facilitate the differential diagnosis of PAH, and to explore the tools for risk assessment in PAH and the risk-management approach to treatment decisions.
PAH causes cardiovascular remodelling and cardiac damage over time ultimately leading to right heart failure, which is the primary cause of death in patients with PAH.1 However, PH starts in the lungs, initially causing dyspnoea during exercise, fatigue, and weakness.2 Approximately 70–85% of patients with PAH are already categorised as WHO functional Class III/IV at diagnosis.3-5 This is unfortunate, as 5-year survival estimates decrease with increasing WHO functional class at diagnosis: 72.2% for Class I, 71.7% for Class II, 60.0% for Class III, and 43.8% for Class IV.6 Early diagnosis is challenging, however, because early symptoms, such as breathlessness, are nonspecific, so referral to a specialist can take 1–2 years.7
Conversely, early diagnosis and management of PAH can improve prognosis. For example, in the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA), which included 1,588 patients, 5-year survival after diagnosis ranged from approximately 77% for low-risk patients, to 53% for intermediate-risk patients, and 33% for high-risk patients.5 Similarly, the French Pulmonary Hypertension Registry (FPHR) reported decreasing transplant-free survival with decreasing numbers of low-risk criteria.8
The Challenging Journey to Pulmonary Arterial Hypertension Diagnosis: Learnings from Two Clinical Cases of Pulmonary Hypertension
The following clinical cases of patients with PH highlight why and how it is important to differentiate between the different PH subtypes as the prognosis, management, and treatment strategies differ by PH group.
Doctor Alessandra Manes
Case One was of a female patient who had been known to have allergic asthma in her teenage years and was diagnosed at age 32 years for asthmatic exacerbation, with mild increase of pulmonary pressures (Group 3 PH). At age 33 years, after progressive worsening of her symptoms, chest tightness, and recurrent exertional syncope, she was re-evaluated by right heart catheterisation (RHC) and diagnosed with idiopathic PAH (IPAH); she was treated with sildenafil 20 mg three times daily, to which macitentan 10 mg/day was added after 2.5 years following worsening symptoms. At age 39 years, she was classified WHO functional Class III and had marked right ventricular (RV) hypertrophy and dilatation. A vasoreactivity test showed that she was acutely vasoreactive; therefore, her treatment was switched to a calcium channel blocker (CCB) (amlodipine 10 mg twice daily). After 6 months, she had improved to WHO functional Class I, her exercise capacity had increased, there was marked reverse remodelling of the RV, and RV function had improved.
This case highlighted the importance of a vasoreactivity test, which is recommended early in the diagnostic process in the European Society of Cardiology (ESC)/ERS guidelines for patients with IPAH, heritable PAH, or drug-induced PAH.2,9 This is important because patients with vasoreactive IPAH who respond to CCB have very good survival rates.
Doctor Isabel Blanco
Case Two was of a male patient aged 70 years old who was diagnosed with chronic obstructive pulmonary disease (COPD) (Global Initiative for Chronic Obstructive Lung Disease [GOLD] criteria 4) at age 56 years. He also had Type 2 diabetes mellitus and systemic hypertension. At age 60 years, he had suspected Group 3 PH (progressive dyspnoea, New York Heart Association [NYHA] functional Class II–III) and was treated with bronchodilators. Eight years later, he was given long-term oxygen therapy as a result of progressive respiratory failure. At age 69 years, there were no changes in right atrial or RV dimensions. His pulmonary haemodynamics were similar at age 70 years to those at age 60 years (including mildly elevated mean pulmonary artery pressure [mPAP] at both timepoints), but his exercise capacity had diminished considerably, likely because of his COPD.
Although this patient had precapillary PH, PAH-specific treatments would not have been beneficial. It is, therefore, important to consider all relevant patient history and complementary tests (for example, echocardiography and pulmonary haemodynamics), and exercise capacity tests are vital for differential diagnosis. More detailed information on the differential diagnosis of PH, and why it is so important, is included in the next section.
Differential Diagnosis of Pulmonary Hypertension: Pearls for the Pulmonologist
Professor Sean Gaine
Symptoms of PH can be nonspecific, commonly including exertion-induced dyspnoea, fatigue, weakness, angina, and syncope.9 Patients with advanced disease can have peripheral oedema and abdominal distension.9 The nonspecific nature of PH symptoms can lead to delays between symptom onset and diagnosis, delays being ≥2 years for 32% of patients in a recent UK study.10 This could potentially be improved through better education of primary and secondary care providers about PH and earlier use of echocardiography in patients with unexplained breathlessness, notably looking at the right-sided chambers of the heart.
It is crucial to establish the correct PH diagnosis, as different forms of PH have different treatment approaches.9 In a patient with suspected PH, the first step is clinical assessment: patient history and physical examination. Then, various diagnostic tests should be undertaken, such as echocardiography, ventilation/perfusion (V/Q) scan, CT, pulmonary function tests, diffusing capacity of the lungs for carbon monoxide (DLCO), blood tests, and RHC.11
The most common type of PH is Group 2 (PH due to left heart disease [LHD]), which accounts for 68% of cases, followed by Group 5 (unclear/multifactorial; 15%), and Group 3 (PH due to lung diseases/hypoxia; 9%).12,13 Therefore, Group 1 (PAH) and Group 4 (PH due to pulmonary artery obstructions, for example, chronic thromboembolic PH [CTEPH]) are the least common, but it is important to identify such patients as there are a range of effective treatments. For Groups 2, 3, and 5, treatments tend to target the underlying disease.
For Group 2 PH (PH-LHD), echocardiography is a key diagnostic tool, and can be used to assess left ventricular (LV) systolic and diastolic dysfunction, and estimate pulmonary artery systolic pressure.14 Several echocardiographic parameters differ between PH-LHD (Group 2) and precapillary PH (Groups 1, 3, 4, 5), so this can help with differential diagnosis.14 Clinical assessment of risk factors for LHD (including hypertension, atrial fibrillation, diabetes, and obesity) can also help to recognise patients in Group 2.14 Pearl 1: an enlarged left atrium is very indicative of PH-LHD.15
Group 3 PH includes obstructive and/or restrictive lung diseases, hypoxia without lung disease (sleep apnoea, for example), and developmental lung disorders.15,16 Chest X-ray can be used to diagnose Group 3 PH by identifying lung diseases such as emphysema and interstitial lung disease.17 It can also show signs indicative of other PH groups, for example, Kerley B lines and pleural effusion in Group 2 PH.18 Pearl 2: a descending right pulmonary artery diameter ≥20 mm is strongly correlated with PH in patients with COPD.19,20 Further, on CT imaging, a main pulmonary artery diameter to ascending aorta diameter ratio ≥1 may predict PH.17,19-21 Contrast CT can be used to visualise right heart dimensions, giving an indication of the severity of RV dysfunction.21 CT can also reveal abnormalities in the lung parenchyma, which can help to discriminate between Groups 1 and 3 PH.17,21 Pearl 3: an RV:LV ratio ≥1.0 is strongly associated with mortality or lung transplantation in patients with Group 3 PH.22 Pulmonary function tests can be used to identify clinically significant obstruction or restriction as risk factors for Group 3 PH. DLCO is reduced in patients with PAH,22,23 and a severely reduced DLCO should raise suspicion for pulmonary veno-occlusive disease, pulmonary capillary haemangiomatosis, or scleroderma-associated PAH.24 Pearl 4: a severely reduced DLCO in IPAH is associated with older age, male sex, smoking history, and poor outcome, and has been termed ‘vanishing capillary syndrome’.24
V/Q scanning is vitally important for the diagnosis of Group 4 PH, as a V/Q mismatched defect (pattern of preserved ventilation and regional absent perfusion) is a key feature of CTEPH.9,24,25 Specific diagnostic signs for CTEPH can also be seen by CT pulmonary angiography, MRI, or conventional pulmonary angiography, including ring-like stenoses, webs, and chronic total occlusions (pouch lesions or tapered lesions).9 Pearl 5: a diagnosis of CTEPH can be made if mPAP is ≥25 mmHg and pulmonary artery wedge pressure ≤15 mmHg after 3 months of effective therapeutic anticoagulation.9 However, mPAP >20 mmHg may be a more appropriate cut-off in the future.9 Group 5 PH includes haematological disorders, metabolic disorders, and sarcoidosis.16 Patients with sarcoidosis can have PH as a result of direct granulomatous involvement of vessels, mediastinal fibrosis, or direct parenchymal involvement.
Group 1 PH (PAH) is partly diagnosed by a process of elimination, i.e., patients with a high probability of PH (by echocardiography), but with a negative V/Q scan, and no clinically significant LHD or lung disease.11 Such patients should be referred to a PH expert centre, where PAH can be confirmed by RHC haemodynamics.9 Of note, patients at high risk of developing PAH (e.g., those with connective tissue disease, HIV, or portal hypertension) should have a fast track referral to an expert centre.11 Pearl 6: markers of RV function (e.g., RA pressure and cardiac index) are more important in determining prognosis than mPAP in patients with PAH.9 Haemodynamics that indicate precapillary PH include mPAP ≥25 mmHg, pulmonary artery wedge pressure ≤15 mmHg, and pulmonary vascular resistance ≥3 Wood units.9 There have been proposals to reduce the threshold for a diagnosis of PH from an mPAP of 25 mmHg to 20 mmHg,12 but this has not yet been agreed in guidelines.
How to Assess Risk and Optimise Treatment in Patients with Pulmonary Arterial Hypertension
Professor Marius Hoeper
Risk assessment, at diagnosis and every 3–6 months during follow-up, is very important for patients with PAH as treatment options (Figure 1) vary depending on whether they are considered to have a low, intermediate, or high risk of 1-year mortality based on clinical evaluation, exercise capacity, and RV function (Table 1).2,9,26 At diagnosis, most patients with PAH are nonvasoreactive and at low/intermediate risk, indicating initial oral combination therapy. Initial monotherapy is generally reserved for select patients: age >75 years, suspicion/high probability of pulmonary veno-occlusive disease, HIV, portal hypertension, very mild disease, or patient responders with IPAH on high-dose CCB.26 Patients who present at high risk should receive initial combination therapy including intravenous prostacyclin analogues.2,9,26
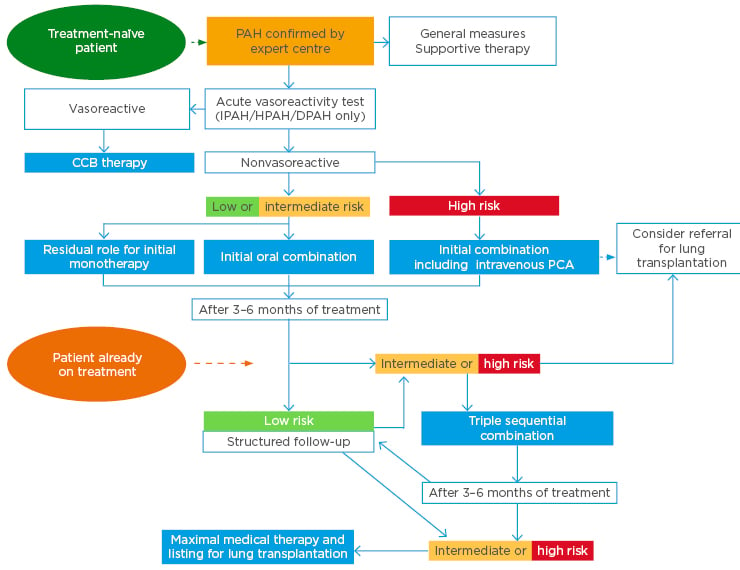
Figure 1: Treatment algorithm for patients with pulmonary arterial hypertension.
CCB: calcium channel blockers; DPAH: drug-induced pulmonary arterial hypertension; HPAH: heritable pulmonary arterial hypertension; IPAH: idiopathic pulmonary arterial hypertension; PAH: pulmonary arterial hypertension; PCA: prostacyclin analogue.
This material has not been reviewed prior to release; therefore the European Respiratory Society may not be responsible for any errors, omissions or inaccuracies, or for any consequences arising there from, in the content. Reproduced with permission of the © ERS 2020. European Respiratory Journal 2019 53: 1801889; DOI: 10.1183/13993003.01889-2018.
Overall risk can be calculated by assigning 1, 2, or 3 points for low, intermediate, or high risk, respectively, for each row of Table 1 and then calculating the average ‘score’. Patients with a mean score of 1.00–1.49, 1.50–2.49, and 2.50–3.00 are then considered to be low, intermediate, or high risk, respectively. This approach was seen in the Swedish PAH Register (SPAHR)27 and the COMPERA registry.5 Both registries showed good discrimination between risk groups for 5-year survival, with similar results (SPAHR: approximately 84%, 52%, and 35%;27 COMPERA: approximately 77%, 53%, and 33%,5 for low-, intermediate-, and high-risk patients, respectively; both p<0.001). The FPHR took a different approach, and categorised patients based on how many low-risk values they had out of four criteria from Table 1 (WHO functional Class, 6-minute walk distance [6MWD], right atrium pressure, and cardiac index).8 They reported decreasing transplant-free survival with decreasing numbers of low-risk criteria (p<0.001).8
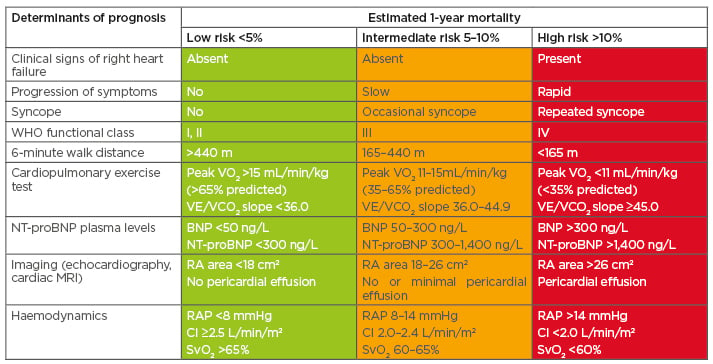
Table 1: Risk assessment in patients with pulmonary arterial hypertension.
BNP: B-type natriuretic peptide; CI: cardiac index; NT-proBNP: N-terminal pro B-type natriuretic peptide; RA: right atrium; RAP: right atrial pressure; SvO2: mixed venous oxygen saturation; VE/VCO2: ventilatory equivalents for carbon dioxide; VO2: oxygen consumption; WHO: World Health Organization.
This material has not been reviewed prior to release; therefore the European Respiratory Society may not be responsible for any errors, omissions or inaccuracies, or for any consequences arising there from, in the content. Reproduced with permission of the © ESC &ERS 2020. European Respiratory Journal Oct 2015, 46 (4) 903-975; DOI: 10.1183/13993003.01032-2015.
Mortality risk can also be assessed using the REVEAL scores.28 REVEAL 2.0 assigns points for 11 weighted clinical parameters (WHO Group I subgroup, demographics, comorbidities, NYHA/WHO functional Class, vital signs, all-cause hospitalisations ≤6 months, 6MWD, B-type natriuretic peptide [BNP], echocardiogram, pulmonary function, and RHC), which are summed to estimate risk.28 REVEAL registry patients at low risk had higher 1-year survival rates than those at intermediate or high risk (approximately 98%, 93%, and 75%, respectively).28 REVEAL Lite 2 includes blood pressure, heart rate, estimated glomerular filtration rate/renal insufficiency, BNP/N-terminal pro B-type natriuretic peptide, NYHA/WHO functional Class, and 6MWD, while REVEAL Lite 1 also includes age, sex, and PAH aetiology.29
For low-/intermediate-risk patients, initial combination therapy is generally recommended over initial monotherapy (Figure 1).2,9,26 This is because the AMBITION study showed that treatment-naïve patients with PAH randomised to initial combination therapy (ambrisentan plus tadalafil) had a 50% lower risk of a morbidity/mortality endpoint than those randomised to initial monotherapy (pooled ambrisentan or tadalafil) (hazard ratio [HR]: 0.50; 95% confidence interval [CI]: 0.35–0.72; p<0.001).30
As PAH is a chronic condition, it is also important to assess risk during treatment (Figure 1).2,9,26 In the SPAHR,27 patients who transitioned from intermediate-/high-risk at diagnosis to low-risk at follow-up had similar 5-year survival to those who were stable low-risk (approximately 96% and 89%, respectively). This indicated that the goal of treatment should be to achieve and maintain a low-risk profile.
The benefit of escalating treatment has been shown in the SERAPHIN31 and GRIPHON studies.32 In the SERAPHIN study, among 308 patients who were already receiving background PAH therapy (mainly [97.4%] a phosphodiesterase-5 inhibitor [PDE-5i]) at baseline and were randomised to placebo or macitentan 10 mg, the addition of macitentan reduced the risk of morbidity/mortality by 38% (HR: 0.62; 95% confidence limit: 0.43–0.89; p=0.009).31
In the GRIPHON study, among 376 patients who were already receiving a PDE-5i and an endothelin receptor antagonist at baseline, the addition of selexipag was associated with a 37% reduction in morbidity/mortality versus placebo (HR: 0.63; 95% CI: 0.44–0.90).32 This beneficial effect was seen in patients with mild symptoms (WHO functional Class II) (64% risk reduction; HR: 0.36; 95% CI: 0.14–0.91) and, albeit to a somewhat lesser extent, those with more advanced symptoms (WHO functional Class III) (26% risk reduction; HR: 0.74; 95% CI: 0.50–1.10).32 Among all patients in GRIPHON (i.e., including 780 who were not receiving a PDE-5i and an endothelin receptor antagonist at baseline), the patients randomised to selexipag were more likely to have improved risk than those randomised to placebo (p<0.001).33 Among all patients, selexipag had a more pronounced treatment effect if it was initiated closer to the time of PAH diagnosis (≤6 months: 55% reduction; HR: 0.45; 95% CI: 0.33–0.63, >6 months: 30% reduction; HR: 0.70; 95% CI: 0.54–0.91; Figure 2),34 highlighting the importance of early intervention.
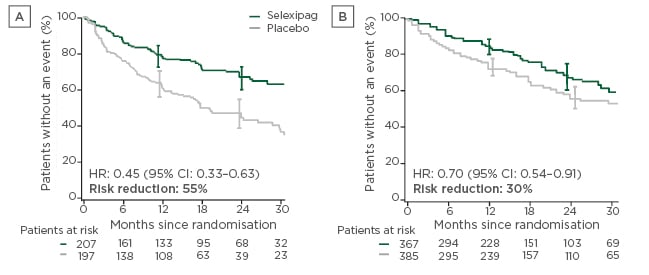
Figure 2: Time from randomisation to first morbidity/mortality event in patients with pulmonary arterial hypertension.
A) Pulmonary arterial hypertension diagnosed ≤6 months before baseline; and B) pulmonary arterial hypertension diagnosed >6 months before baseline.
CI: confidence interval; HR: hazard ratio.
Adapted with permission from Gaine et al.34
Closing Remarks
Professor Marion Delcroix
Regarding differential diagnosis, clinical symptoms and physical signs of PH can be difficult to distinguish in patients with respiratory disorders;9 however, establishing a correct diagnosis of PH and the specific group are crucial in order to tailor treatment.9 Multiparametric risk assessments should be undertaken at diagnosis and 3–6-monthly during follow-up to assess disease severity, predict survival, and guide treatment decisions.9,26 Assessment of risk changes over time is also essential for making treatment decisions. Most low-/intermediate-risk patients with PAH should receive initial double-combination therapy,9,26 with early escalation to triple-therapy in those not at low risk within the first 3–6 months26 as targeting multiple pathological pathways in PAH is key to improving patient outcomes.
Date of preparation: November 2020
EM-46407