Meeting Summary
The symposium explored the use of state-of-the-art technologies in building the evidence for Alpha 1 Antitrypsin (AAT) efficacy, namely the use of computed tomography (CT) and recent developments in regional lung density analysis, and current challenges and data gaps for the management of AAT deficiency (AATD). The vital importance of registries in building our knowledge and understanding of AATD and its management were also discussed. Dr Stolk opened the symposium with a brief overview of AATD and the results of the RAPID clinical trial programme, which provided evidence for the efficacy of AAT therapy in slowing the rate of lung density loss in AATD. Prof Parr then presented the rationale and methodology for assessing regional lung density changes, as measured by CT, and the potential clinical relevance of regional treatment variability in AATD. Prof Ficker addressed the clinical implications for AATD treatment, in light of data from the RAPID clinical trial programme, and provided an overview of the current challenges for treating patients with AATD, including questions surrounding when to commence AAT therapy and how the potential life-extending effect of AAT therapy can be assessed and quantified. Finally, the importance of registries was discussed; Prof Strange provided an overview of the USA Alpha 1 Foundation registry and presented key published data. In addition, he discussed current and future initiatives. Dr Stolk considered the European Alpha 1 International Registry (AIR) and presented the results of recent projects supported by this registry.
Welcome and Introduction
Doctor Jan Stolk
AATD, a hereditary disorder characterised by reduced levels of functional AAT, results in excess lung protease activity and increases the risk for development of chronic obstructive pulmonary disease (COPD). Patients present with early-onset emphysema characterised by loss of lung density and is more prominent in the lower lobes. Lung densitometry assessed by CT scanning has been used to measure disease progression and demonstrate treatment efficacy in clinical trials of AAT.
Results of the RAPID clinical trial programme, consisting of RAPID-RCT followed by the open- label extension RAPID-OLE, provided evidence for the efficacy of Respreeza® in slowing the rate of lung density loss (Figure 1). RAPID-RCT, which randomised 180 patients with AATD and forced expiratory volume in 1 second (FEV1) 35–70% predicted, to either Respreeza (n=93; intravenous AAT 60 mg/kg body weight per week) or placebo (n=87), showed that the annual rate of lung density decline at total lung capacity was significantly reduced in the Respreeza group (-1.45 g/L/year) compared to the placebo group (-2.19 g/L/year; p=0.03).1
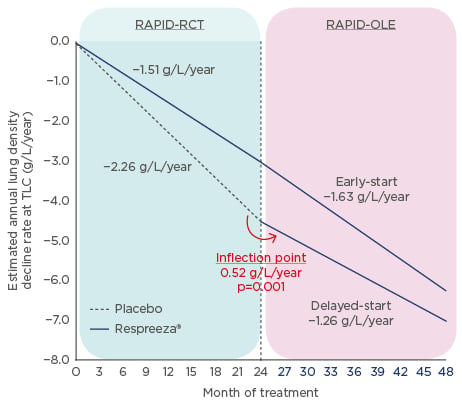
Figure 1: RAPID clinical trial programme results.
Annual lung density decline rate of whole lung densitometry (volume-adjusted PD15) at total lung capacity during 48 months of treatment (completer population).
OLE: open label extension study; RCT: randomised controlled trial; TLC: total lung capacity.
Adapted from McElvaney et al.2
Patients who received either Respreeza (n=76) or placebo (n=64) in RAPID-RCT were then included in the 2-year open-label extension trial, RAPID-OLE.2 Results showed that between Months 24 and 48, the rate of lung density loss was reduced in the delayed-start group compared to placebo in Months 0–24.
This means that patients who initially received placebo during RAPID-RCT experienced a disease- modifying reduction in the progression of emphysema (from -2.26 g/L to -1.26 g/L per year) that was similar to that of actively treated patients. This slowing of lung density loss was highly statistically significant (p=0.001). For the early-start group, (i.e. patients who had received Respreeza during RAPID-RCT), no significant difference was seen over 4 years (-1.51g/L/year to -1.63 g/L/year), and their treatment effect was maintained during this time period. However, some questions still remain, including how CT changes relate to clinical outcomes and how registries can support efforts to improve treatment.
Regional Lung Density and Imaging: Latest Findings from the RAPID Extension Trial
Professor David Parr
Prof Parr opened his presentation by describing the principles of CT lung densitometry, explaining that CT scanners are tissue densitometers that produce images comprising pixels corresponding to a volume of lung tissue, hence the term voxel. Each voxel has a computed value of density relative to water measured in Hounsfield units (HU); the density of water is 0 HU, air is -1,000 HU, and normal lung density is between -550 HU and -950 HU. Emphysema results in tissue loss and hyperinflation that leads to loss of lung density that is generally accepted as abnormal when it falls below -950 HU. CT densitometry has been validated as a measure of emphysema against pathology and a variety of clinical parameters. In AATD, two pivotal studies have shown that loss of CT lung density correlates with decline in health status over time3 and with decline in FEV1.4
The EXACTLE study was an exploratory study of the use of whole lung CT densitometry to assess therapeutic effects of AAT therapy and was not powered to show a treatment effect.5 However, in a post hoc analysis of EXACTLE data, Parr et al.6 showed that differences in treatment effect between patients receiving AAT therapy and those given placebo were evident when the basal region of the lung was sampled (p=0.040), but not in the central (p=0.155) and apical (p=0.673) regions.6 Similar results were reported in a post hoc analysis of data from the RAPID clinical trial programme;7,8 the greatest treatment effects (i.e. difference between placebo and AAT therapy) occurred in the basal lung regions.
Prof Parr went on to explore potential explanations for these observations. A histopathological study from the 1970s9 investigating whole lung specimens from patients with COPD showed that centrilobular emphysema was more frequently located towards the apex of the lung, while panlobular emphysema was found towards the base. Furthermore, a study from the 1990s showed the coefficient of variation of interalveolar wall distance was significantly higher for the same mean linear intercept in centrilobular emphysema than panlobular emphysema.10 Such data suggest distinct pathological subtypes of emphysema.4
These two histopathological subtypes of emphysema also coexist in patients with AATD; about one-third of patients with panlobular emphysema due to AATD also have centrilobular emphysema evident on CT,11 and the pattern of emphysema distribution also influences the physiological impairment. In a quantitative CT scan study of two groups of patients with either predominantly basal or apical emphysema, basal distribution was associated with relatively greater impairment of airflow but less impairment of gas exchange than apical distribution.12 Such data suggest that centrilobular and panlobular emphysema are distinct pathological subtypes with potentially different pathogenesis. However, regional lung density changes on CT in patients with emphysema may arise from regional volume changes or from tissue loss. Consequently, interpretation of regional density changes in a group of patients may be misleading and further studies are required to characterise the natural history of emphysema progression in individual patients.
Clinical Implications and Challenges for the Treatment of Alpha 1 Antitrypsin Deficiency
Professor Joachim H. Ficker
A meta-analysis of five classical studies,13 including 1,509 patients, demonstrated that patients with AATD receiving AAT therapy show less decline in FEV1 (Figure 2).
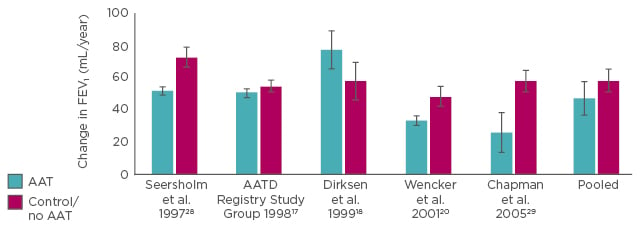
Figure 2: Effect of Alpha 1 Antitrypsin therapy on forced expiratory volume in 1 second.
Meta-analysis: the pooled results imply that therapy with AAT slows the loss of lung function.13
AAT: Alpha 1 Antitrypsin; AATD: Alpha 1 Antitrypsin deficiency; FEV1: forced expiratory volume in 1 second.
FEV1 is a measure of bronchial obstruction that reflects many different pathological changes and, as such, is a poor predictor of the degree and progression of emphysema. However, for patients with AATD, the primary aim is to reduce progression of emphysema and researchers need to understand how to best measure this; therefore, it is important to understand how FEV1 changes relate to emphysema progression.
It has been shown that expiratory air flow limitation in emphysema is, in part, due to the loss of elastic recoil.14 During the development of emphysema, bronchial tethering, which secures patency of the airways, is lost, leading to a checked valve phenomenon of dynamic airway collapse. This can be visualised on CT scans by the different forms of the bronchi during inspiration and expiration, and relates to a characteristically deformed flow-volume loop and reduced FEV1.15 Such data indicate that FEV1 is not a direct measurement of emphysema, but represents an indirect measurement of airway collapse due to loss of elastic recoil.
In addition, the decline in FEV1 found in patients with AATD is not linear. A study of 100 patients with AATD, grouped according to baseline FEV1 (mild, moderate, severe, and very severe disease), showed FEV1 decline over 3 years was greatest for those with moderate disease.16 In mild disease, loss of elastic recoil does not reach the threshold required for bronchial collapse and in severe disease there is nearly total airway collapse and emphysema progression causes no further loss of FEV1. In line with this, there is little change in FEV1 in both early-stage and late-stage disease, with time during long-term follow-up indicating that FEV1 is an insensitive measure of emphysema progression in comparison to CT lung density.17,18
Using change in lung density as a primary endpoint, the RAPID clinical trial programme demonstrated that emphysema progression can be significantly reduced by AAT replacement therapy.1,2 Long-term changes in lung density were similar in all degrees of lung function impairment and active treatment was associated with a significant lung density preservation (p<0.001 between placebo and active treatments) independent of baseline FEV1.19
A post hoc analysis of data from the RAPID clinical trial programme2 suggests that patients receiving AAT therapy will achieve critical lung densities (average final recorded lung density of approximately 21 g/L), where terminal events such as death or lung transplantation occur around 5.6 years later than those patients not receiving treatment (Figure 3).
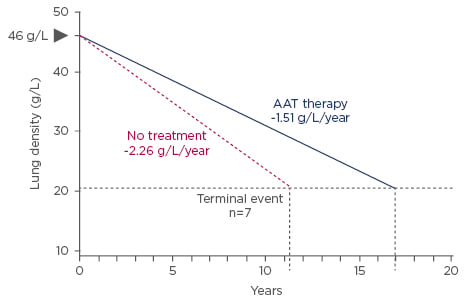
Figure 3: Time to respiratory crisis.
Earlier treatment start may result in a more pronounced effect.
AAT: Alpha 1 Antitrypsin.
Adapted from McElvaney et al.2
Such extrapolation suggests that differences in lung density decline might ultimately translate into overall survival differences. Therefore, early introduction of treatment in all patients with emphysema-related AATD may delay time to respiratory crisis.
Whether different AATD phenotypes should be handled in different ways needs to be addressed. Data suggest that AAT therapy provides a particular benefit in arresting disease progression in patients with rapidly deteriorating emphysema.2,20 Furthermore, data from the Danish AATD registry showed a marked difference in survival between index (those who present with symptoms) and non-index (those diagnosed via a family member) cases, with a mean age of death of 49.4 years for index cases versus 69.3 years for non-index cases.21
In addition to rapid decliners and index versus non-index cases, other AATD phenotypes include genotypes with intermediate AAT levels and rare alleles with very low levels (PIZ [Null], and PI [Null, Null]). A paper by Ferrarotti et al.22 reviewed 6,057 individuals enrolled in the Swiss Cohort Study on Air Pollution (SAPALDIA) and showed that AAT serum levels vary according to different genotype classes in the general population, and that certain allele combinations of the SERPINA1 gene (PI*SZ, PI*ZZ, and Null/Null) increase the risk of emphysema. These data suggest that rather than basing AAT therapy dose on body weight alone, additional phenotypic characteristics might have to be considered when choosing the optimal dose for treatment. In line with this hypothesis, patients may need higher doses during exacerbations. Planned and ongoing studies will investigate the currently licensed dose of 60 mg/kg body weight against the efficacy of higher doses of AAT therapy.
It is important to introduce outcomes into clinical trials that matter to patients. To date, the only patient-centred outcomes used have been hospital admissions (EXACTLE study)5 and extrapolating reductions in lung density decline to mortality (RAPID clinical trial programme).2 However, it is difficult to recruit sufficient numbers of patients into clinical trials of rare diseases such as AATD, and long-term observational studies based on registries may be more practical than traditional randomised trials for gathering data on patient-centred outcomes and may offer additional insights.
Going Beyond the Trials: Strengthening Clinical Evidence
Professor Charlie Strange
With 7,000 different rare diseases affecting 10% of the population, understanding the course of the disease and the development of treatments may be a daunting task, particularly if the number of affected individuals is very small. Registries can be an important research tool that can also facilitate recruitment to clinical trials.
One of the best examples of a registry is the USA Cystic Fibrosis Foundation Registry, which tracks the health outcomes of roughly 28,000 patients with cystic fibrosis each year, producing a snapshot of the therapies received and overall health outcomes. The foundation, which employs >50 people, also manages the largest cystic fibrosis clinical trials network in the world.
Membership of the USA Alpha 1 Foundation registry increased from 500 members in 1998 to 6,416 individuals in 2017. The rapid growth can be attributed, in part, to a USA primary care initiative that tested patients with COPD for AATD. Significant publications have come from the registry, and include a study of individuals with severe AATD showing that, in addition to cigarette smoking, male sex, previous asthma, chronic bronchitis, and pneumonia diagnoses represent risk factors for severe COPD.23
The USA Alpha 1 Foundation is currently developing a new registry that will include information on genotype, diagnostic history, FEV1 category, lung function, outcomes, therapies, visit type, and vitals, as well as a biorepository for samples. The registry will also contain an integrated informatics for biology at the bedside tool (known as i2b2) to enable exploratory analysis of the registry. It will also include a facility to allow the larger Alpha 1 community, such as patients and researchers, to review anonymised data upon request. Additionally, the Alpha 1 Global community is attempting to synchronise registry fields around the world to enable shared data collection and the gathering of a larger database. There is a need to focus on long-term outcomes, including quality of life and mortality.
Greater emphasis is being placed on patient engagement, for example with the work of the Patient-Centred Outcomes Research Institute (PCORI). This trust fund receives $150 million per year and has the mission to examine the relative health outcomes, clinical effectiveness, and appropriateness of different medical treatments. Patient engagement is important because it helps improve patient recruitment and retention rates, patient understanding of results, and enhances trust between researchers and participants. A 2013 PCORI survey revealed that barriers to engagement from the patient perspective include lack of time (43%), concerns about privacy (36%), and work, school, or care giving commitments (33%), while facilitators included helping others with their medical condition (68%), learning about their health (63%), helping the next generation (57%), and getting paid (56%). From the clinician’s perspective, barriers included lack of time (79%), payment (47%), and research training (35%), while facilitators included helping patients receive better care (79%), getting paid (78%), and contributing to scientific knowledge (61%).24
Going forward, it will be important to make as much data public as possible, and use political will to integrate as many data sources as possible.
Clinical Implications and Challenges for Treatment of Alpha 1 Antitrypsin Deficiency
Doctor Jan Stolk
More than 10 European Union (EU) member states currently contribute data to the European AIR. The registry was founded in 1996 by chest physicians following publication of a World Health Organization (WHO) report on AATD.25 Data of individuals with genotypes ZZ, SZ, or rare variants are being considered. The minimal data set includes information about demography, smoking habits, primary reason for AAT analysis (e.g. respiratory symptoms or family screening), medical history, information about AAT therapy, spirometry, and qualitative information on liver enzymes. Soon after AIR was started in 1997, it had 540 subjects, and after a rapid period of expansion the numbers reached 5,389 subjects in 2016.
AIR data show the median age of diagnosis is similar for men and women (46.54 years and 46.65 years, respectively) and similar for ZZ, SZ, and rare genotypes (46.39 years, 48.97 years, and 46.68 years, respectively). With regard to phenotype, 84.6% of participants are ZZ (n=4,388), 11.9% SZ (n=617), and 3.5% rare variants (n=179), of which the greatest contribution comes from patients with a Null/Null genotype. Additional registry data show a median age of respiratory symptom onset of 40 years, differing for the three subgroups (40 years for ZZ, 45 years for SZ, and 39 years for rare variants). On average there was a 7-year delay between onset of respiratory symptoms and AATD diagnosis. A USA study showed that 44% of patients reported seeing at least three physicians before an initial diagnosis was made.26
The registry also showed that quality of life, as assessed by the St George’s Respiratory Questionnaire (SGRQ) at baseline, was worse for smokers and ex-smokers compared to never-smokers and index cases versus non-index cases, respectively.27 The registry has investigated a subpopulation of patients with one of the 32 AAT Null mutations currently known, which result in an absence of AAT in the serum.27 A matched analysis of Null AATD patients and ZZ and SZ patients from the AIR registry (based on age and sex) showed Null patients have significantly lower lung function values than SZ and ZZ individuals (p=0.001 for both FEV1 and the coefficient of diffusion of carbon monoxide).27
In order to investigate the effect of treatment with AAT in this very small population of patients with Null mutations, the registry compared patients with a minimum of 6 years of follow-up on treatment in Italy, Spain, or France (n=10) with those who had received no treatment in the Netherlands due to lack of reimbursement (n=12). Results showed an average annual loss of FEV1 for untreated patients of 76 mL/year versus 12 mL/year for treated patients. This suggests that in selected populations with ultra-rare mutations, AAT therapy protects against FEV1 decline. It would be impossible to explore such therapy in a randomised trial due to the rarity of the phenotype.
The AIR registry has provided a rich source of data for analysis of the clinical characteristics of AATD. However, quality control of data represents a particular challenge.
Summary
Doctor Berend Stoel
Providing an overview of the symposium, Dr Stoel said that it has taken 25 years from the first clinical study that utilised CT densitometry to measure emphysema progression in AATD until the publication of the RAPID study demonstrating that AAT therapy slows lung tissue destruction. CT densitometry is a sensitive method for assessing lung tissue but has downsides, including exposure to X-rays and the need for stringent quality control. In future, it is hoped that more specific blood biomarkers that are sensitive to changes in disease severity and progression will become available to replace CT scanning.
Alpha 1 registries represent real-world clinical practice. They can be used for recruiting patients to clinical trials and describing natural progression of disease in different phenotypes. Identification of phenotypes that benefit most from therapy could help define the patient groups who should be eligible for AAT therapy reimbursement.
Question and Answer Session
Q: How should we approach the evaluation of survival without treatment? What clinical outcome markers should be used and are these affordable for registries?
A: Considering how to approach the issue of patients who do not receive AAT therapy, Prof Strange said the National Heart, Lung, and Blood Institute (NHLBI) registry enrolled patients with and without AAT therapy over 5 years and had shown a mortality signal. However, the finding was downplayed because it was not derived from a prospective randomised trial. The signal was strong and it would have taken only 2 years of treatment to show this mortality difference. A caveat, he said, was that mortality largely occurred in patients with FEV1 <30% predicted at baseline, making it important to perform studies in the most severely affected patients. However, with the associated comorbidities in these patients this may not be possible. In future, the USA registry plans to collect death certificates for cause of death and also declines in lung function.
Q: Should biomarker data, similar to that found in the RAPID study relating to elastin degradation, be used as an endpoint in future clinical studies?
A: The RAPID study demonstrated that elastin degradation breakdown products, desmosine (DES) and isodesmosine (IDES), were significantly reduced following AAT treatment. Furthermore, levels of DES/IDES could be related to CT density. Prof Parr said studies demonstrate placebo patients experience greater loss of lung mass than patients receiving AAT therapy. A complication, however, is that disease progression causes hyperexpansion of the lungs, which reduces density even in the absence of a change in lung mass. Observations of changes in both lung mass and elastin biomarkers suggest changes occur at the tissue level.
Q: Would studies focussing on data from lower lobe lung segments allow fewer patients to be enrolled and shorter follow-up periods?
A: Prof Parr stated that, while statistical data is better in the lower part of the lung, investigators should still monitor the entire lung. This is because it is impossible to judge whether any density changes occurring in the lower lung are due to a real loss of lung density or due to changes in other parts of the lung. Such limited data, he felt, would be unlikely to convince regulators.
Q: Should guidelines be changed to allow treatment to be continued even in patients with very low FEV1 % predicted?
A: Prof Ficker commented that AAT therapy should be started as early as possible to preserve lung structure, and he did not see any reason to stop treatment when the patient declined. The situation, he said, was analogous to osteoporosis, where treatments are started as soon as possible to preserve bone mass.
Q: What new data could be used to persuade healthcare payers to support reimbursement of AAT therapy?
A: Regarding healthcare payer data, Prof Strange felt that it would be valuable to have data on the effects of AAT therapy on exacerbation frequency as this influences healthcare utilisation and cost. It was important, he added, for databases to capture real-life patient experiences, including exacerbations.
Prof Ficker underlined differences in Europe, where AAT therapy is not reimbursed in Scandinavia and the UK. One of the aims of the European Reference Network for Rare Diseases, he said, was to harmonise treatment reimbursement across EU member states.
Q: Where in the field of COPD is the best data on patient exacerbations?
A: Prof Strange answered that the Alpha 1 Foundation runs a programme called Alpha Net, which asks patients to answer an Alpha Net exacerbation questionnaire every month. The scheme, which has been running for 10 years, has allowed real-world data to be captured that is only just starting to be mined.
Q: Would investigators be ready to enrol AATD subjects with normal lung function into placebo-controlled trials?
A: Prof Parr commented that treating patients with normal lung function was fine; however, it would be hard to justify if they did not show emphysema on CT scans, as they were not considered at-risk. Prof Strange felt that in gene therapy trials it would be justifiable for patients with normal lung function to be considered as candidates for therapy. Prof Ficker said that all alpha 1 patients showing evidence of emphysema should be treated.