
Abstract
Occupational exposure to indoor air moulds and the consequent development of dampness and mould hypersensitivity syndrome (DMHS) may cause lung damage; in most cases, this is not allergic asthma mediated by specific immunoglobulin E-class antibodies. Instead, it is often a hypersensitivity pneumonitis or bronchopneumonitis. In Finland, the current diagnostic criteria for occupational DMHS have been adapted from knowledge of immunoglobulin E-mediated asthma; however, the safety of the methods used in occupational medicine have been insufficiently addressed in the literature. Accordingly, the aim of this paper is to raise awareness about the safety of current methods: specific inhalation challenge, workplace peak expiratory flow monitoring, and histamine provocation tests, by illustrating four cases. The medical records of these four cases with documented occupational DMHS were reviewed. The presented evidence suggests that the methods applied to study the occupational nature of lung damage are not suitable and the current ethics are questionable. The authors claim that, in particular, serial inhalation challenge with extracts from moulds, workplace serial peak expiratory flow leading to continuous exposure to mycotoxins, and histamine provocation tests may irreversibly damage the health of DMHS patients. Therefore, there is a prompt need to revise current practice guidelines to assess occupational DMHS. The guidelines should not be based on old dogmas, nor should they be influenced by insurance considerations. Instead, they should be based solely on medical evidence and, crucially, they should be safe for the patient and, therefore, should be implemented with caution.
INTRODUCTION
Mould-related disease, or dampness and mould hypersensitivity syndrome (DMHS), has been extensively described;1-9 it is a complex multiorgan disorder with activation or impairment of the immune system,10,11 systemic inflammation,12,13 recurrent infections, or reactivation of latent infections. Only some cases develop into immunoglobulin (Ig)E-mediated allergy and asthma,14,15 and DMHS may also be associated with invasive fungal infections.16 In reality, DMHS is a mycotoxicosis,17,18 a systemic chronic inflammation,12,13 and an oxidative stress reaction.17 The differences in response patterns between patients have been reviewed through the prism of evolutionary coadaptation of moulds and humans over millennia of coexistence.19
DMHS is very common in Finland, although exact data on the number of incidences are missing. The diagnostic coding R68.81 implemented in Finland in 2015 does not refer specifically to DMHS, but includes all environmental hypersensitivities. DMHS is therefore considered as a trait or a functional disorder, not a disease, and does not guarantee the patient access to any social security benefits. In Finland, the incidence of DMHS in adults and children is increasing alarmingly and is highlighted in media publications and on social networks. However, officially recognised occupational cases, mainly occupational asthma (OA), are steadily decreasing. Only 5–6% of DMHS patients receive compensation for OA (Irmeli Lindström, unpublished data, 2016) and DMHS is accepted as an occupational disorder only if it results in the development of asthma; all other forms of DMHS12 are not considered.20,21 This health policy is determined by the Finnish Institute of Occupation Health (FIOH), which is partly financed by insurance companies.22
From 1995 to 2009, the FIOH studied the causality of OA and mould infestation in the workplace by applying the specific inhalation challenge (SIC); this was mandatory for all subjects undergoing investigations for occupational lung conditions.21 This exposure was performed without ethical approval. During the SIC, an extract of Aspergillus fumigatus or Cladosporium cladosporioides (ALK-Abelló, Copenhagen, Denmark) was inhaled by a sensitised person in a specific chamber. Importantly, these preparations contained impurities23 and had never been intended for inhalation but only to study IgE-mediated immunity. Fungal preparations were also administered to individuals without specific IgE-class antibodies (Cases 1 and 2). During this period, the SIC test was performed on several hundreds of people, some of whom became unconscious after the exposure, and many experienced acute health deterioration (Cases 1 and 2), requiring hospitalisation. After the SIC exposure, some of these individuals were diagnosed with allergic alveolitis (AA) (Cases 1 and 2). After many complaints, the SIC was replaced by workplace serial peak expiratory flow (PEF) monitoring, which became mandatory.21 To the best of the authors’ knowledge, the safety of these tests has not been addressed in the scientific literature. This case series reviews the medical records of four patients with an explicit occupational exposure to dampness microbiota documented by state-of-the-art environmental investigations.
CASE 1
The Specific Inhalation Challenge Test Evoked an Acute Neutrophilic and Lymphocyte Influx into the Pulmonary Alveoli
A 50-year-old non-smoker experienced dizziness and a feverish feeling at his workplace; he had previously had a massive exposure with an unconscious episode due to indoor air moulds while standing below a ventilation output in October 2000. The replacement air in the remediated building was taken from under the floor. Extensive growth of Streptomyces bacteria, along with other damp-related species, had been cultured prior to remediation. In 2001, the man was placed on sick leave and thereafter did not return to his workplace. He was referred to the FIOH, where he tested IgE-negative for all available mould antigens and IgG-positive for some fungal antigens, but not for A. fumigatus. In March 2002, the first SIC was performed with A. fumigatus and was repeated after 1.5 weeks. The patient felt feverish and fatigued after the A. fumigatus exposure but did not react to the C. cladosporioides and Acremonium exposures; altogether he was exposed to SIC four times over 12 days. A few days after the first exposure to A. fumigatus, he underwent his first bronchoalveolar lavage (BAL) investigation, which was repeated in July 2002. During the bronchoscopy, the mucosa was found to be fragile and covered with bloodstains. Finally, in 2002, he was given a diagnosis of occupational AA and received a disability pension for several years. After SIC exposure, he experienced vertigo, felt feverish although his body temperature remained at only 36°C, had shortness of breath, and his walking ability declined dramatically. In February 2013, he was admitted to hospital due to the presence of right pleural exudate that was treated with pleural decortication. The patient continues to experience vertigo and sick building syndrome, and a mouldy environment exacerbates his symptoms (e.g., pain). In summary, the patient in this case was exposed to dampness microbiota at his workplace; he had four sequential SIC tests and immediate BAL investigation revealed acute inflammation, leading to a diagnosis of occupational AA.
CASE 2
The Specific Inhalation Challenge Test Exacerbated Pulmonary Effusion in Dampness and Mould Hypersensitivity Syndrome
A 49-year-old non-smoker worked in an office with dampness in 1990 and 4 years later started to experience recurrent sinusitis. Starting from 2000, she experienced unexplained bruising and, in 2001, moved to another building because moisture damage microbiota (e.g., Chaetomium and Aspergillus) and asbestos had been found. In this new office, she started to experience a non-productive cough, fatigue, dyspnoea, palm tingling, and fever. In 2001, she had leukopenia and thrombocytopenia and, due to high fever, she was admitted to hospital; however, pneumonia was not diagnosed. Thereafter, she often missed work due to illness, which improved her condition. In 2002, she was referred to the FIOH, where skin prick tests for moulds were negative, as were IgE-class antibodies to 16 common damp microbiota moulds. High-resolution computed tomography (HRCT) revealed minor fibrosis, and a histamine provocation test in 2002 confirmed asthma. Subsequently, she was given a 3-month sick leave period, was prescribed asthma medication, and was then able to cycle 20–30 km per day. She returned to work in a third building and soon afterwards was again referred to the FIOH where spirometry showed evidence of obstruction. She experienced pain in her chest when sneezing and in the evenings she was hypothermic. She was again referred to the FIOH where she was exposed to SIC tests with A. fumigatus and C. cladosporioides extracts under powerful corticosteroid medication with A. fumigatus antigen and C. cladosporioide antigen in 2003. Thereafter, she had a burning and seizure-like sensation in her chest, tingling of her left arm, mouth numbness, and a heavy feeling beneath the scapulae. The SIC results were interpreted as an intrinsic but poorly balanced asthma. She became sensitised to environmental moulds and could not tolerate damp weather. She developed multiple allergies, including to bananas, strawberries, and apples. She returned to work at the end of 2002, but was soon placed on sick leave again. In 2003 (1 year after SIC), she underwent BAL and ultimately a diagnosis of occupational AA was made. In 2004, she was examined again at the FIOH, where her condition worsened, and she needed oxygen inhalation. In 2006, a BAL examination was performed again that confirmed her previous diagnosis. In 2009, she underwent biopsy of her lungs that caused massive oedema of her neck. In 2014, the BAL examination again confirmed AA but insurance companies have refused to compensate her medical expenses since 2014. In summary, the patient described in Case 2 was exposed to dampness microbiota in multiple workplaces, where serial PEF monitoring was performed two times with inconclusive results. She was exposed once to SIC and BAL investigations that revealed chronic inflammation compatible with AA.
CASE 3
Mandatory Serial Peak Expiratory Flow Monitoring for Legal Evidence Caused a Health Deterioration
A healthy 50-year-old non-smoker started to feel unwell shortly after she moved to a new office in January 2009. She had severe flu-like symptoms with cough, rhinitis, eye infections, palpitations in the extremities, loss of voice, headache, fatigue, and fever. During her time off work in the summer, her symptoms eased, but when she returned to work in August she started to experience dyspnoea. In addition, she began to experience nausea, vomiting, and chest pain. Species, including Actinomycetes, Acremonium, and Penicillium, were detected at her workplace. In December 2009, she was placed on sick leave due to new-onset asthma and she was referred to the FIOH for professional evaluation. The FIOH insisted on serial PEF monitoring, although she felt unwell in the office. The return to work aggravated her illness during workplace PEF monitoring and she was subsequently admitted to hospital. The attending physician doubted her ability to continue PEF monitoring, but nonetheless its completion was deemed mandatory in order to gather evidence of an occupational illness. In March 2010, regular medication for asthma was started. In June 2010, she received a diagnosis of OA and a pension for her professional disability for only 2 years; the reasoning for this decision was that other diseases, such as multiple chemical sensitivity (MCS), were her main ailments. At present, she has chronic fatigue syndrome, hyperhaemoglobinaemia (haemoglobin: 170 g/L; reference for females: 117–155 g/L), secondary (compensatory to chronic toxicosis) polycythaemia, and hypokalaemia (plasma potassium: 3.1–3.2 mmol/L; reference: 3.3–4.9 mmol/L) despite potassium substitution. In 2017, a disturbance in her autonomic nervous system balance was documented (Figure 1). Due to her MCS, she found it difficult to leave her home and rarely had visitors. At the time of this communication, she is not a recipient of any social security benefits. In summary, the patient in Case 3 was exposed to dampness microbiota at her workplace; she undertook workplace serial PEF twice, despite her health deterioration, and finally received a diagnosis of OA.
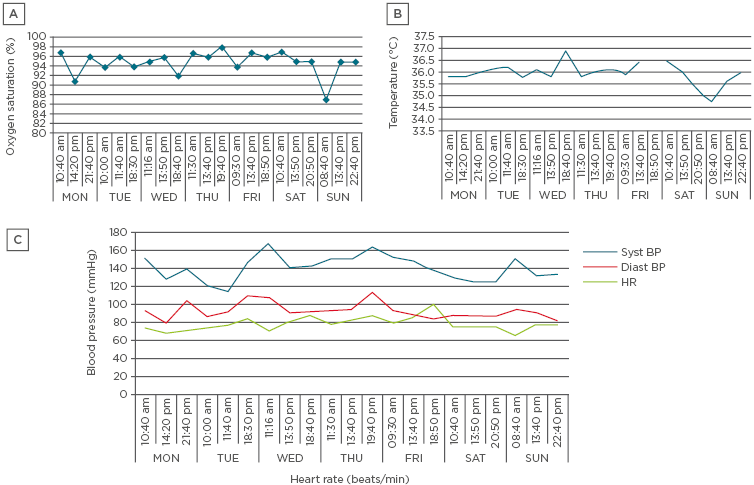
Figure 1: Physiometry parameters indicating dysregulation of the autonomic nervous system.
A) Monitoring of oxygen saturation (Jumper Medical Equipment Co., Shenzhen, China); B) Body temperature (Beurer, Ulm, Germany); C) Monitoring of systolic and diastolic blood pressure, and heart rate. P-glucose was also monitored (Bayer, Leverkusen, Germany) but not recorded in this figure. During the episodes of hypoglycaemia or hypothermia, the patient felt so unwell that she was bedridden.
Diast BP: diastolic blood pressure; HR: heart rate; syst BP: systolic blood pressure.
CASE 4
Controlled Significant Decline of Peak Expiratory Flow During a Bronchospasm is of No Legal Value
A 58-year-old non-smoker started to experience a non-productive cough at her workplace when studying petri dishes with bacterial growth. She started to experience flu-like episodes in the workplace after the weekends. She was previously healthy, taking no medication. In June 2014, after a holiday, she received two antibiotic courses for sinusitis that did not relieve her symptoms. She lost her voice and her cough became so intense that she experienced pain in her chest and ribs and felt extremely fatigued. During a 2-week period of sick leave, her voice almost returned to normal but her cough continued and worsened after her return to work. Finally, a fungal growth was found in the proximity of her office, and it was no longer disputed that her disease was associated with working in the office. In August 2014, she underwent six maxillary punctures, but the cultures were negative. The workplace serial PEF in August was unsuccessful due to abundant secretions of mucus from her nose, shortness of breath, and pain in her ribs and chest. From mid-August to December, she was placed on sick leave due to severe cough and laboratory remediation.
At the FIOH, in January 2015, incipient asthma was suspected because of the variation in the daily PEF, the insignificant response to bronchodilators, and slight hyper-reactivity, but these findings did not meet the clinical criteria for asthma. She returned to work in January 2015 when remediation of the laboratory had been completed; she quickly started to experience a loss of voice and her cough exacerbated. On her last working day, when she experienced a bronchospasm, her PEF measurements dropped to 280–280–300 L/min (normal value: 450–500 L/min; recorded by a nurse) and 4 hours later, after 2 hours of outdoor walking, her PEF had slightly recovered (360–370–360 L/min; recorded by a physician). A diagnosis of asthma was made in August 2015 at the Helsinki University Hospital, Helsinki, Finland. Lung HRCT in 2016 revealed multiple lymphatic nodules in the interstitia that were still present 6 months later. Since her serial PEF had been unsuccessful, it was deemed that she did not have OA and thus did not qualify for benefits. Cladosporium, Penicillium, and Aspergillus versicolor were cultured in her workplace. In summary, the patient presented in Case 4 was exposed to dampness microbiota; workplace serial PEF were not performed but a significant drop in PEF on her last working day was of no legal value with regard to government benefits.
DISCUSSION
These cases illustrate that the diagnostic methods and criteria24 applied to an IgE-mediated immune response are being erroneously applied to a disease that, in most cases, is not IgE-mediated (Cases 1 and 2).15 Medical and legal abuse of patients exposed to indoor air dampness microbiota in their workplaces continues to take place in Finland with the unspoken approval of all the appropriate monitoring authorities. The Declaration of Helsinki,25 signed in 1964 and widely regarded as the cornerstone document on human research ethics, is being violated.
The authors have shown that patients developed sick building syndrome (Cases 2, 3, and 4), meaning that while being away from the workplace their condition improved, but worsened upon return. It has also been illustrated that DMHS is mostly not an IgE-mediated allergy; none of the cases reported here were IgE-positive to the most common fungal antigens. DMHS patients may develop a loss of tolerance, becoming sensitive to allergens they could tolerate before (Case 2), and exposure to an inhaled fungi antigen caused acute inflammation (Case 1), while a BAL test performed 1 year after SIC revealed chronic inflammation (Case 2). Both Cases 1 and 2 illustrate the deleterious health effects of SIC. These cases show that DMHS may also associate with MCS (Case 3). Most importantly, continued exposure to mycotoxins endangers patients’ health (Cases 2, 3, and 4) and workplace serial PEF may be inconclusive (Case 2), may be of no help for the patient’s legal rights (Case 3), and may aggravate symptoms (Case 3).
On the basis of the presented data, the authors conclude the following:
Deduction 1
Exposure to indoor air moulds may cause lung damage; in most cases, this is not an allergic asthma mediated by specific IgE-class antibodies, but is AA or hypersensitivity pneumonitis (HP).26 DMHS has a myriad of clinical presentations.1-9 Lung effusion due to the inhalation of spores and mycotoxins often is not only asthma; instead, in the majority of cases, it is AA or HP.26 It is the opinion of the authors that the criteria and the protocol devised for OA27,28 should not be applied to DMHS. The recommendations that an individual who has lost their tolerance should continue to inhale toxic air endangers their health, disregards the consequential symptoms and conflicts with the universally accepted healthcare principle of primum non nocere (first, to do no harm). Gathering legal evidence should never take precedence over medical ethics.
Mycotoxins cause so-called ion channel disease by forming novel ion channels that disrupt the membrane potential of the mitochondria because of the influx of Na+ and efflux of K+ from the cell.18 Mycotoxins are broad-spectrum toxins with cytotoxic and immunomodulatory effects.5-7,18 Chronic exposure to moulds may induce an inflammatory response that can be measured by cytokine and chemokine production from peripheral blood mononuclear cells.29 Long-term exposure to indoor air dampness microbiota is the foundation for the development of MCS (Case 3).9 One may argue that the reported consequences of mycotoxin exposure refer to the oral administration route; however, mycotoxins are also absorbed via the inhalation route, after which they can gain access to the blood circulation without being detoxified in the enterohepatic circulation, or alternatively they can penetrate directly into the brain via the nervus olfactorius.30
Since inhaled particles such as spores of pathogenic indoor moulds are only 0.005–5.000 µm in diameter,31 it is easy to comprehend that these xenobiotics penetrate deep into the lungs, creating inflammation in situ, not only airway hyper-reactivity and inflammation. When a post-mortem examination was performed on an individual who had inhaled large quantities of mould xenobiotics through a bagpipe, a severe HP was revealed.26 Thus, inhalation of mould components may lead not only to the inflammation of the large airways (asthma), but to an overwhelming inflammation of the parenchyma26 and small airways.32 When both are present, this condition may be called bronchopneumonitis (BP). Moreover, the strict definition of asthma has been questioned.32 There are several reasons to suspect that mould exposure is not primarily allergic asthma but in fact causes HP or BP: a) patients report a poor response to bronchodilators because the inflammation is mainly in the small airways or in the parenchyma; b) poor response to corticosteroids because of the involvement of the T helper 17 inflammatory cell arm;33 c) during auscultation, wheeze is not predominant in mould-exposed individuals and, instead, shortness of breath and even chest pain at rest are usually reported; d) spirometry curves are often compatible with a restriction defect rather than with an obstruction pattern; e) HRCT may reveal lymphatic nodes in the interstitial parenchyma (Case 4) or incipient fibrosis (Case 2); and f) an influx of lymphocytes, with the typical ratios of their subsets, is compatible with AA (Case 2).34 Immediately after the exposure to impure mould extracts, a pathology compatible with acute inflammation (neutrophilic influx) was documented (Case 1).
Deduction 2
Clinical criteria and the protocol for evaluating mould-related lung disease in DMHS should be revised. Exposure to wet mouldy grains is not the only reason an individual can develop HP or BP. Thus, it is not only a farmer’s disease and is not synonymous with organic dust toxic syndrome (ODTS).35 In ODTS, the exposure is massive and caused mainly by spores, whereas the exposure to damp microbiota is associated with mycotoxins and volatile organic compounds. Indoor mycotoxins may be different from outdoor mycotoxins;18 indoor mycotoxins have been demonstrated to inhibit the growth and function of antigen-presenting cells and lymphocytes.11,12 Therefore, AA, HP, or BP due to indoor mycotoxin-producing moulds may develop, even though there are low lymphocytic cell counts. Thus, the criteria adopted for diagnosing occupational AA associated with DMHS should be different from those of farmer’s disease or ODTS. AA (or HP or BP depending on the agreed terminology) should be examined appropriately in every patient exposed to dampness microbiota.36
Deduction 3
The SIC test is by no means the most accurate test27,28 to study causality in OA, especially in DMHS. The test is invasive and may cause irreversible health damage (Cases 1 and 2). As performed in Finland, the SIC test has been responsible for many serious and under-reported health problems in DMHS patients. The large number of SIC tests performed in Europe28 is, in fact, a shameful history, not an achievement of advanced occupational medicine.
Deduction 4
Workplace serial PEF monitoring to prove causality in DMHS should be discontinued. Workplace serial PEF was originally suggested as a way of assessing OA with positive specific IgE-class antibodies.24 IgG and IgE-class antibodies to dampness moulds have been extensively studied in Finland. It was found that specific IgE elevation to 11 species of moulds was observed in <5% of exposed children attending problematic schools (approximately n=500; age: 7–13 years).15 The majority of IgE-positive children were atopic. Moreover, the PEF test has a sensitivity of only 75% (specificity: 95%),24 which is insufficient for screening purposes. Rather, the possibilities of using cytokine and chemokine measures of blood in the diagnosis of asthma caused by mould exposure should be considered.29 PEF measurement per se is not harmful, but serial workplace PEF measurements will cause continued harmful exposure of a person to indoor air mycotoxins (Case 3) and therefore should be banned.
Deduction 5
Histamine provocation tests in the evaluation of hyper-reactivity of bronchi in DMHS patients should be abandoned. So far, there is no evidence about the safety of this intervention. Many patients with DMHS in whom MCS has developed9 exhibit a disruption in the permeability of their blood brain barrier (BBB). Iatrogenic exposure to histamine that penetrates the BBB will aggravate inflammation in the brain. In DMHS patients, neuroinflammation recorded as a structural brain injury with increased permeability of the BBB has been documented.37
CONCLUSION
Finally, it is undisputable that DMHS is not primarily an invasive fungal disease. Therefore, immunity guidelines developed for invasive infection are not applicable to this clinical entity.38,39 The authors argue that DMHS is primarily a mycotoxicosis. Evaluation of the SIC test40-43 shows that it lacks safety considerations and a careful assessment by independent clinicians of medical ethics, and may cause possible long-term adverse effects and even iatrogenic damage. The future directions for diagnosing and treating HP with an incidence of 0.3–0.9 per 100,000, irrespective of its cause, were highlighted by Vasakova et al.44
Based on the presented arguments, the authors challenge current practices related to the interpretation of occupational DMHS. The causality should be proven with a safer technique; for example, assaying the biomarkers of the inflammation cascade and oxidative stress.44 These biomarkers should have a short half-life but be stable enough to permit analysis. The possibilities are within our reach; we need goodwill and an open mind to improve our practices.





