Abstract
Adalimumab is a recombinant fully human monoclonal antibody targeting soluble and transmembrane TNF alpha. It is approved for the treatment of immune-mediated rheumatic, gastroenterological, dermatological, and ophthalmological conditions and this therapeutic versatility has made it the top-selling drug worldwide since 2012. Not surprisingly, following the patent expiration of the originator drug, biopharmaceutical companies invested in the development of biosimilar versions of adalimumab and six have already received marketing authorisation: ABP 501, GP2017, and BI 695501 in Europe and in the USA (though the manufacturer of the latter requested authorisation withdrawal in Europe), and SB5, FKB327, and MSB11022 in Europe. This manuscript reviews published data on approved adalimumab biosimilars, including analytical and biofunctional results from preclinical assessments; pharmacokinetics after administration in healthy subjects (Phase I trials); and efficacy, safety, and immunogenicity from pivotal (Phase III) clinical trials. Data on switching from reference adalimumab to biosimilars, and predicted cost-savings from available budget impact models, will also be addressed.
Introduction
Adalimumab is a recombinant fully human monoclonal antibody (IgG1 type) targeting soluble and transmembrane TNF. AbbVie’s bio-originator adalimumab, branded name Humira® (AbbVie, USA), is the top global selling drug since 20121 and it is approved for the treatment of immune-mediated inflammatory conditions of rheumatic, ophthalmological, dermatological, and gastroenterological nature. Adalimumab is indicated for rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis, juvenile idiopathic arthritis (polyarticular and enthesitis-related arthritis), psoriasis, hidradenitis suppurativa, adult and paediatric Crohn’s disease, ulcerative colitis, and adult non-infectious uveitis. In the European Union (EU), but not in the USA, adalimumab is also indicated for non-radiographic axial spondyloarthritis, paediatric psoriasis, paediatric hidradenitis suppurativa, and paediatric non-infectious uveitis.2
The approaching date of patent expiration, alongside the prospect of entering a several billion-dollar market, has led biopharmaceutical manufacturers to invest in the development of biosimilar versions of adalimumab. By the time this manuscript was elaborated, six biosimilars were given positive opinions by the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA): ABP 501 (Amgevita®, Solymbic®, Amjevita®, Amgen, USA), GP2017 (Hefiya®, Halimatoz®, Hyrimoz®, Sandoz, Germany), and BI 695501 (Cyltezo®, Boehringer Ingelheim, Germany) in Europe and in the USA, and SB5 (Imraldi®, Biogen, South Korea), FKB327 (Hulio®, Fujifilm Kyowa Kirin, Japan), and MSB11022 (Idacio® and Kromeya®, Fresenius Kabi, Germany) only in Europe.3,4 Table 1 summarises adalimumab biosimilars already approved and currently being developed in highly regulated markets. In the USA, litigation between AbbVie and adalimumab biosimilar manufacturers over adalimumab’s patent was resolved by a settlement that protects the patent until 2023.5 In Europe, the patent expired in October 2018 and the first biosimilars have recently entered the market. It should be noted, however, that the manufacturer of BI 695501 requested withdrawal of marketing authorisation in Europe due to unresolved patent litigation with AbbVie in the USA.6

Table 1: Adalimumab biosimilars already approved and currently being developed in highly regulated markets.3,4
*Marketing authorisation withdrawn by the manufacturer.
The current article performs a comprehensive review of adalimumab biosimilars approved in highly regulated markets, including available preclinical data, pharmacokinetics (PK), efficacy, safety, and immunogenicity assessments, and pharmacoeconomic considerations.
Preclinical Development of Adalimumab Biosimilars
The stepwise, totality-of-evidence development of any biosimilar product has its mainstay in the demonstration of a high degree of similarity in analytical and biofunctional evaluations between the biosimilar candidate and reference product. After this extensive preclinical phase, an abbreviated clinical phase ensues, including the assessment of PK, efficacy, safety, and immunogenicity.7
Adalimumab biosimilar candidates were tested against several batches of USA and Europe-sourced reference products for key quality attributes such as primary structure (molecular mass, protein sequence, and post-translational modifications), high-order (secondary and tertiary) structures, product-related and host-cell impurities, general properties, and product stability. State-of-the-art, sensitive, and orthogonal analytical methods were employed, many of them developed or adapted specifically for this purpose. Batches of biosimilar candidates were compared with reference products using pre-established similarity ranges or direct side-by-side comparisons.
After a thorough assessment of preclinical data within the marketing authorisation application, the regulatory agencies considered there was sufficient information to ensure a similar clinical performance in ABP 501, SB5, GP2017, BI 695501, FKB327, and MSB11022, despite some minor quality differences found in some of these candidates, which were duly justified and were not expected to impact PK, efficacy, or safety.8-13 For instance, SB5 had a slightly higher amount of free sulfhydryl groups, as well as charged N-glycans and acidic variants compared to reference adalimumab.9 FKB327, on the other hand, showed differences in the glycosylation profile, with higher mannose content.12 This difference led inclusively to further in vitro bioassay testing and PK data statistical reanalysis, before similarity was confirmed.12
Due to the pleiotropic nature of TNF, all known adalimumab mechanisms of action with potential clinical relevance must be compared in vitro. Furthermore, biofunctional testing also demonstrates that differences in quality attributes, should they exist (for instance, post-translational modifications), do not impact in vitro biological activity. Biofunctional data provided by ABP 501, SB5, GP2017, BI 695501, FKB327, and MSB11022 manufacturers showed a high degree of similarity in both Fab and Fc-mediated functions, including, but not limited to, binding and neutralisation of soluble and transmembrane TNF; binding to FcRn, FcγRIa, FcγRIIa, and FcγRIIIa receptors; and antibody-dependent and complement-dependent cytotoxicity.9,11-15
Although not mandatory, most of these biosimilar manufacturers provided PK and toxicology assessments in animal models in the data package presented to the regulatory agencies, once again demonstrating a high degree of similarity to reference products.
Clinical Performance of Adalimumab Biosimilars
All biosimilar candidates demonstrated PK equivalence with EU and USA-sourced reference adalimumab in Phase I trials performed in healthy subjects, with the confidence intervals of the primary endpoints (area under the curve [AUC] and maximum drug concentration [Cmax]) falling within the prespecified range of 0.80–1.25 (Table 2).16-21 In accordance with regulatory requirements, Phase III trials were performed using a randomised, double-blind, parallel-group design. RA was chosen as the disease population in all but MSB11022, which was tested in patients with plaque-type psoriasis; ABP 501 and GP2017 also have available studies in this condition (Table 3). All biosimilar candidates confirmed similar efficacy, safety, and immunogenicity to reference adalimumab.18,22-28
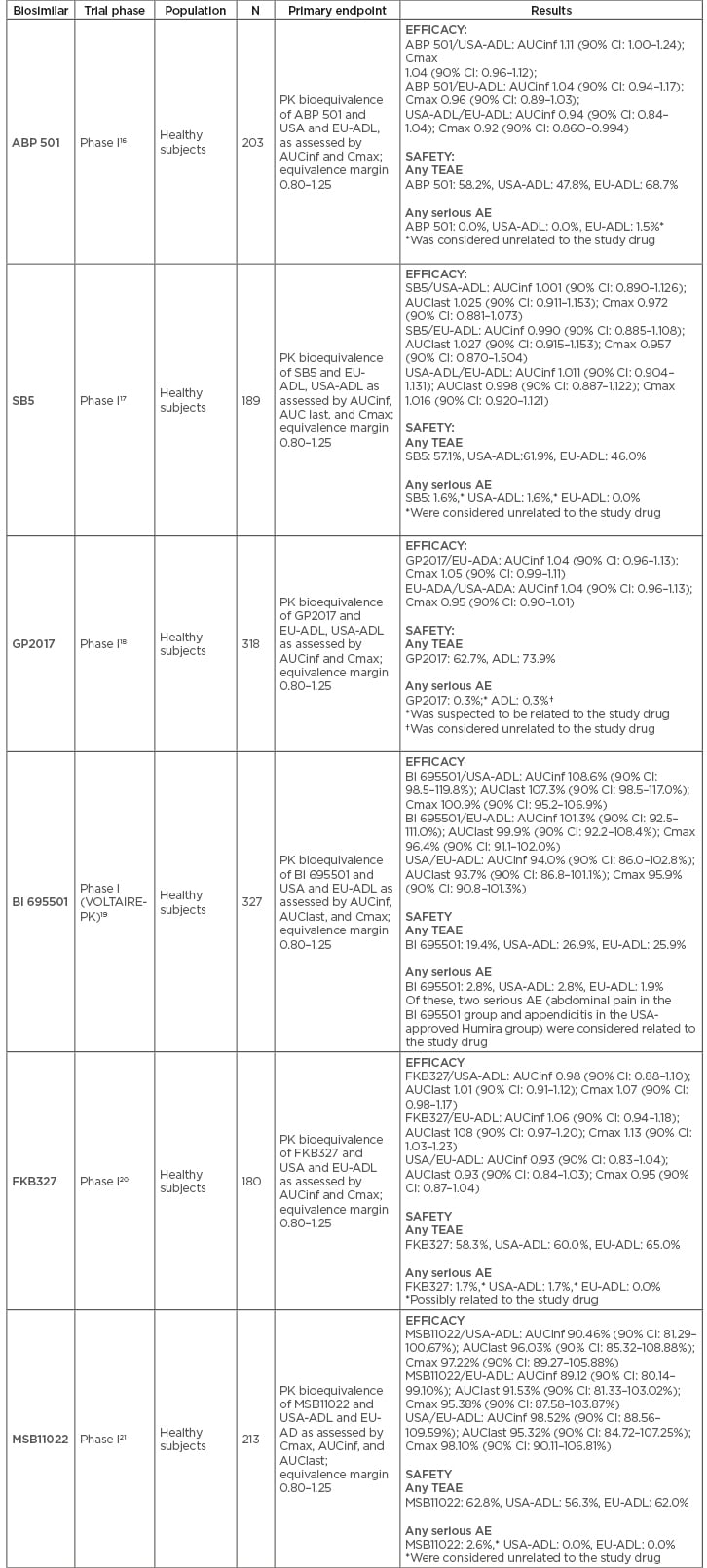
Table 2: Phase I clinical trials for each approved adalimumab biosimilar.
AE: adverse event; AUCinf: concentration time curve (AUC) from time 0 extrapolated to infinity; AUClast: concentration time curve (AUC) from time 0 extrapolated to last quantifiable concentration; CI: confidence interval; Cmax: maximum (peak) serum concentration; EU-ADL: European Union-sourced adalimumab; PK: pharmacokinetic; TEAE: treatment emergent adverse event; USA-ADL: United States of America-sourced adalimumab.
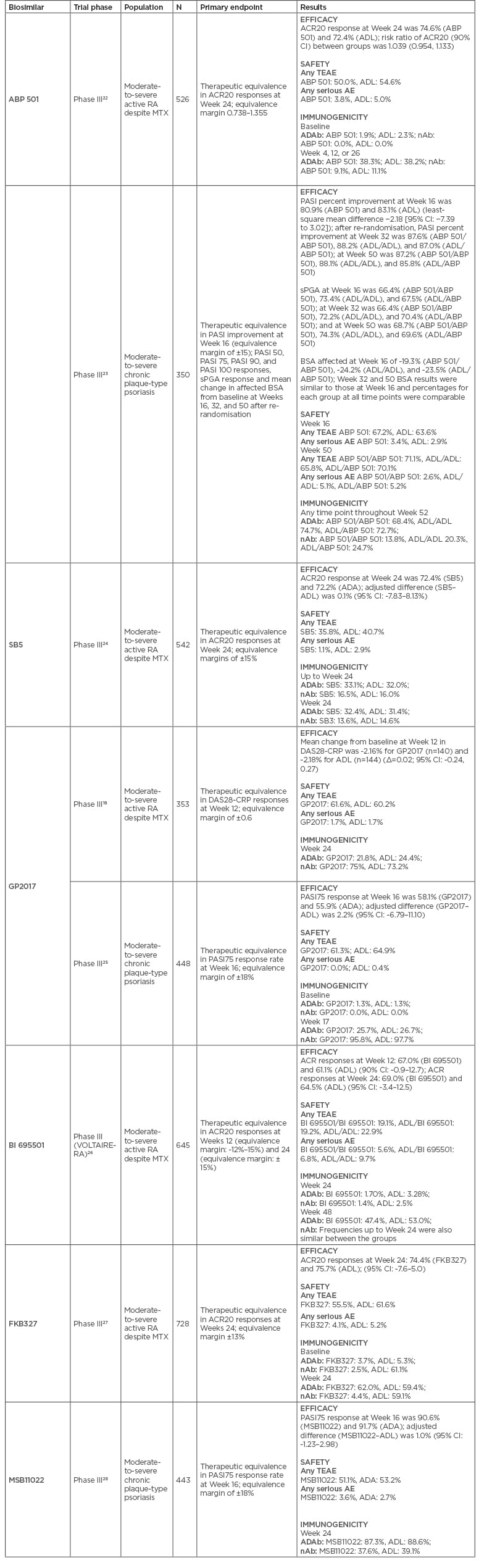
Table 3: Phase III clinical trials for each approved adalimumab biosimilar.
ACR20: American College of Rheumatology 20% improvement criteria; ADAb: antidrug antibody; ADL: adalimumab; AE: adverse event; BSA: body surface area; CI: confidence interval; DAS28-CRP: disease activity score-28 including high-sensitivity C-reactive protein; MTX: methotrexate; nAb: neutralising antibody; PASI: Psoriasis Area and Severity Index; RA: rheumatoid arthritis; sPGA: static Physician’s Global Assessment; TEAE: treatment emergent adverse event.
Efficacy
In Phase III trials, biosimilar candidates must demonstrate equivalence to their reference drug, in contrast with pivotal trials of bio-originators in which superiority over placebo is the endpoint. From a statistical point of view, this means that the confidence intervals of the primary efficacy endpoint(s) must be contained within prespecified equivalence margins that are calculated for each biosimilar drug based on historical data from the reference product and by comparison with prior study designs.29
Primary efficacy endpoints were met in all Phase III trials in RA, namely similar American College of Rheumatology 20% (ACR20) improvement criteria responses at Week 24 between reference adalimumab and ABP 501,22 SB5,24 BI 695501,26 and FKB327,27 and similar mean change in disease activity score-28 including high-sensitivity C-reactive protein (DAS28-CRP) at Week 12 for GP2017 (Table 3).18 Noteworthy, ABP 501 presented statistically significant superiority over reference drug in ACR20 responses at Weeks 2 and 12, but not at other time points or secondary efficacy endpoints.30,31 This was not considered by the regulatory agencies to compromise biosimilarity and was attributed to chance. Secondary efficacy endpoints, including ACR50/70 responses, European League Against Rheumatism (EULAR) criteria responses, and DAS28 variations and remission were also similar between reference drug and biosimilar candidates.18,22,24,26-28 Interestingly, in pivotal trials of adalimumab biosimilars, a slightly higher proportion of patients in both biosimilar and reference arms achieved the ACR20 primary endpoint compared to the active arm in pivotal trials of originator adalimumab, which may be attributed to different trial designs.32
MSB11022 performed similarly to reference drug in Psoriasis Area and Severity Index (PASI) 75 response at Week 16 (primary endpoint) in a Phase III trial in plaque-type psoriasis, confirming biosimilarity.28 This biosimilar was recently assessed in a Phase III trial of RA patients but results were not available to this date. ABP 501 and GP2017 also had Phase III trials in plaque-type psoriasis, showing comparable results in primary (PASI percent improvement and PASI 75, respectively) and secondary endpoints at Week 16.23,25
Despite being assessed in trials of patients with RA and psoriasis, approved adalimumab biosimilars were granted by the regulatory agencies with the remaining clinical indications of the originator drug (extrapolation of indications).
Safety
No new adverse events were found in Phase III clinical trials beyond those expected for the population and drug class, and the majority were classified as mild-to-moderate in severity.
The rate of treatment-emergent adverse events (TEAE) was similar in the biosimilar and reference drug groups, ranging from 19.1–61.6% for biosimilars (ABP 501: 50.0%, SB5: 35.8%, GP2017: 61.6%, BI 695501: 19.1%, FKB327: 55.5%, MSB11022: 58.0%) and 40.7–62.0% for reference adalimumab.18,22,24,26-28 The rate of severe adverse events was also similar in both groups, ranging from 1.1–5.6% for the biosimilar group (ABP 501: 3.8%, SB5: 1.1%, GP2017: 1.7%, BI 695501: 5.6%, FKB327: 4.1%, MSB11022: 3.6%) and 1.7–5.0% for the reference drug group.18,22,24,26-28
Despite similar safety profiles, some minor differences are worth mentioning. For instance, ABP 501, BI 695501, and SB5 had fewer injection site reactions compared to reference adalimumab,22,24,26 which was considered not relevant and attributed to differences in excipients by regulatory agencies. BI 695501 showed increased incidence of analytical changes like anaemia (most in patients with low haemoglobin levels at baseline); bone fractures (but incidence within expected range in the general population); and positive screening for tuberculosis (no active cases). EMA accepted these events as rare and attributed to chance.33
Immunogenicity
The use of a biologic agent can trigger an immune response, which may result in reduced efficacy, treatment failure, or adverse effects.34 Detailed immunogenicity evaluations are required for approval of biosimilars and the types of assays and sensitivity of detection are described in updated regulatory guidance documents.35,36 In the case of rheumatic diseases, 25 studies with immunogenicity data for 16 biosimilars or biosimilar candidates are published: 7 with adalimumab as the reference product (biosimilars BI 695501, SB5, ABP 501, FKB327, and MSB11022, and biosimilar candidates PF-06410293 and ZRC-3197).
Studies of adalimumab in both healthy volunteers and patients varied in methodology of antidrug antibodies (ADAb)/neutralising antibodies (nAb) detection, as well as study design and duration, meaning that comparisons between studies are not a reliable means to determine which biosimilar is more prone to elicit an immune response. Nevertheless, immunogenicity results of Phase III trials are summarised in Table 3.
The incidences of ADAb in adalimumab trials generally increased with trial duration (reaching a plateau after 12–24 weeks of treatment), a phenomenon that was not observed in trials of etanercept, rituximab, and their biosimilars. Typically, ADAb-positive individuals had lower drug concentrations and higher clearance rates compared with ADAb-negative individuals, with effects comparable between reference products and biosimilars. Overall, in adalimumab trials there is evidence that the formation of ADAb is associated with deterioration in certain pharmacodynamic parameters such as CRP or erythrocyte sedimentation rate and diminished clinical efficacy and safety, but the statistical significance of those differences was generally not examined in individual trials.
Cross-reactivity assessments show the ability of ADAb to bind both the reference and biosimilar products and have been reported in only four randomised control trials in rheumatic diseases, one of them with adalimumab and its biosimilar FKB327.20
Biosimilars can be introduced into patients’ treatment regimens, which may affect immunogenicity. Available data for the biosimilars of adalimumab indicate that switching resulted in no changes in quantitative or qualitative immunogenicity (see below). Overall, the ranges of ADAb incidences in pivotal randomised control trials of reference products are lower than those reported in recent trials comparing them to their biosimilars,37 which may be a result of improvements in assay methodology (including sample handling, drug trough levels, validation techniques, sample storage, number of replicates), sensitivity (currently mandated by regulatory agencies),35,36 as well as patient disease status and the trial design employed.38
Switch
All adalimumab biosimilars have information on at least one switch, except MSB11022. Their Phase III clinical trials were extended to a later period of evaluation where patients on the reference drug were re-randomised to switch to biosimilar or to remain on reference drug. These studies had on average a post-switch period of 28 weeks (ABP 501 [psoriasis]: Week 16 to 52; GP2017 [RA]: Week 24 to 48; BI 695501: Week 24 to 48; SB 5: Week 24 to 52; FKB327: Week 28 to 48). ACR20/50/70 response rates and mean change from baseline in DAS28-erythrocyte sedimentation rate were similar across the switched and the continuous groups.18,22-27Also, there were no differences in the rate of treatment discontinuation between groups. In the study of SB5, radiographic results were also analysed. Radiographic progression was comparable between all treatment groups over the course of 52 weeks, and consistent with historical data for reference adalimumab.39 Whilst these studies assessed a single transition from reference to biosimilar drug, GP2017 had a Phase III trial performed in patients with chronic plaque-type psoriasis including a multiple-switch period.25 Patients achieving a ≥50% PASI improvement were re-randomised to maintain their originally assigned treatment or to receive either GP2017 or reference adalimumab during three alternating 6-week periods. Once again, no significant difference in efficacy, safety, or immunogenicity was found between switchers and non-switchers.25
With regard to safety, the different clinical trials also showed similar results between switch and maintenance groups. The rate of TEAE was similar in these groups, ranging from 15.6–54.6% for switch groups (adalimumab to SB5: 37.6%’ adalimumab to GP2017 [RA]: 45.5%, adalimumab to BI 695501: 42.5%; adalimumab to FKB327: 54.6%; adalimumab to ABP5 01 [psoriasis]: 15.6%; adalimumab to GP2017 [psoriasis]: 46.0%), 19.0–55.9% for the adalimumab maintenance groups, and 23.0–53.1% for the biosimilar maintenance groups. The rate of severe adverse events was also similar, ranging from 0.0–5.7% for the switch groups (adalimumab to SB5: 3.2%; adalimumab to GP2017 [RA]: 5.7%; adalimumab to BI 695501: 4.1%; adalimumab to FKB327: 2.8%, adalimumab to ABP 501 [psoriasis]: 0.0%; adalimumab to GP2017 [psoriasis]: 6.0%), 0.0–6.3% for the adalimumab maintenance groups, and 1.3–4.0% for the biosimilar maintenance groups. No hypersensitivity to adalimumab was reported upon switching.18,22-27
Switch data on adalimumab biosimilars are reassuring but should be interpreted with caution, as most trials assessed a single transition in a small number of patients with limited follow-up periods. Further evidence from pharmacovigilance programmes and real-world studies will be necessary to properly assess interchangeability of adalimumab biosimilars.
Pharmacoeconomics of Adalimumab Biosimilars
The bio-originator adalimumab has proven efficacy and safety in the treatment of rheumatic, ophthalmic, dermatological, and gastroenterological conditions. This therapeutic versatility has made adalimumab the top-selling drug worldwide since 2012.1 In 2017 alone, sales reached $18.43 billion for all clinical indications.1 Naturally, expectations are high for the potential cost savings from adalimumab biosimilars and their role in the reduction of the economic burden of biotherapies.
A recently published article by Aladul et al.40 assessed the effect of the introduction of infliximab, etanercept, and adalimumab biosimilars in rheumatology and gastroenterology specialities on UK healthcare budget. The budget impact model built for adalimumab assumed a 33% price discount for the biosimilar drug in the first year and an annual 15% discount up to the fourth year, as well as price erosion of the reference drug reaching 50% at this latter time point. From 2017 to 2020, considering an annual growing market share of 10%, 35%, 60%, and 90%, savings due to Solymbic® (Amgen, USA), Amgevita® (Amgen, USA), and Imraldi® (Biogen, South Korea) are expected to reach £177 million and £91 million in rheumatic and inflammatory bowel diseases, respectively.40 Two other analyses were published as abstracts. The first used data from a USA claims-base and estimated annual combined savings of $6.1 million per 10,000 insured RA patients treated with infliximab or adalimumab biosimilars (assuming a 30% market share and 25% price discount).41 The second estimated combined savings over 1 year of €26 million, €351 million, and €98 million in France, Germany, and the UK, respectively, with the use of infliximab, etanercept, or adalimumab biosimilars in RA patients (assuming a 50% biosimilar quota and 30% price discount).42
Overall, budget impact analyses of adalimumab biosimilars are still scarce, especially when compared to infliximab and etanercept biosimilars. Based on what is known from these therapies, cost savings generated by adalimumab biosimilars will allow thousands of new patients to be treated every year (assuming these savings are re-invested) and will play an important role in the sustainability of healthcare systems worldwide.43
Conclusion
All currently approved adalimumab biosimilars have demonstrated equivalence to reference product in preclinical and clinical studies and have been rigorously scrutinised by regulatory agencies before approval. Further reassurance on the safety and efficacy of adalimumab biosimilars in clinically studied and extrapolated indications will come from mandatory long-term pharmacovigilance programmes established by the FDA and EMA, as well as real-world data from national patient registries. The worldwide success of bio-originator adalimumab will grant its biosimilars an economic impact that will surpass the one seen with infliximab, etanercept, or rituximab, further contributing to the treatment sustainability of patients with immune-mediated inflammatory conditions.