Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease characterised by a breakdown in immune tolerance that induces an attack on normal tissues by the immune system. The dysfunction within both the innate and adaptive immune systems increases cytokine production, B lymphocytic overproduction of autoantibodies, and T lymphocyte activity. Cytokines and inflammatory mediators have been associated with several clinical endpoints, including the activity of disease and outcomes. In fact, some of them have been associated with different clinical subphenotypes (e.g., lupus nephritis), suggesting their role as biomarkers, and, in some cases, therapeutic targets. Thus, knowledge of the pathophysiological processes associated with the development of SLE could aid in setting up better diagnostic and therapeutic approaches to reduce the high burden of disease, and thus improve quality of life and outcomes. Herein, the authors have compiled a concise review of the clinically relevant cytokines and inflammatory mediators associated with SLE and its manifestations.
INTRODUCTION
Systemic lupus erythematosus (SLE) is the prototypical autoimmune disease (AD), with a prevalence of up to 178 per 100,000 habitants, with an increased incidence and severity in non-Caucasian patients.1 Patients typically show burdensome symptoms of psychological distress and a marked effect on physical function that impairs their quality of life.2 Clinical manifestations include arthritis, rash, serositis, cytopenia, kidney disease, and neurological involvement.3
SLE is characterised by the dysfunction of both the innate and adaptive immune systems, which increase the production of cytokines and other inflammatory mediators (Figure 1).4 These molecules produce a strengthening of inflammatory responses, an increase in the apoptosis of circulating cells, a defect in clearing apoptotic bodies, and an overproduction of autoantibodies, which are associated with diverse clinical subphenotypes. New biomarkers, such as soluble urokinase plasminogen activator receptor (suPAR), osteopontin, and soluble Fas (sFas), have arisen as valuable tools to measure activity and severity of disease.5-7 However, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels remain the most useful tools to evaluate activity of disease.8
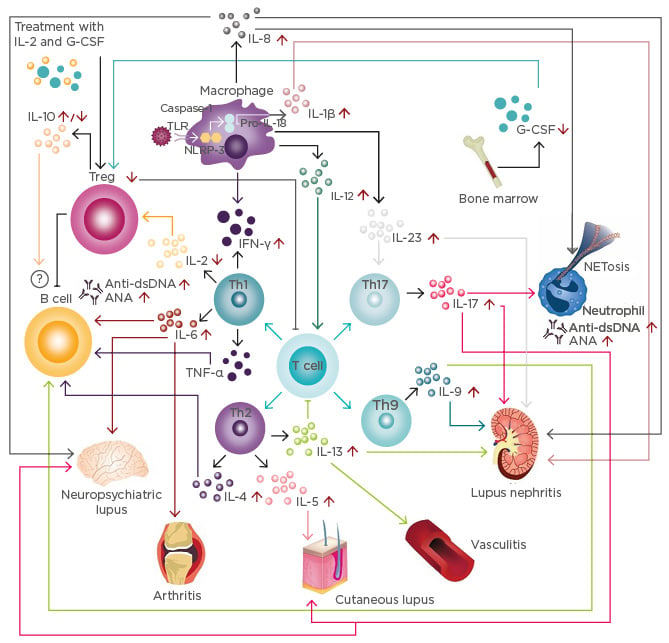
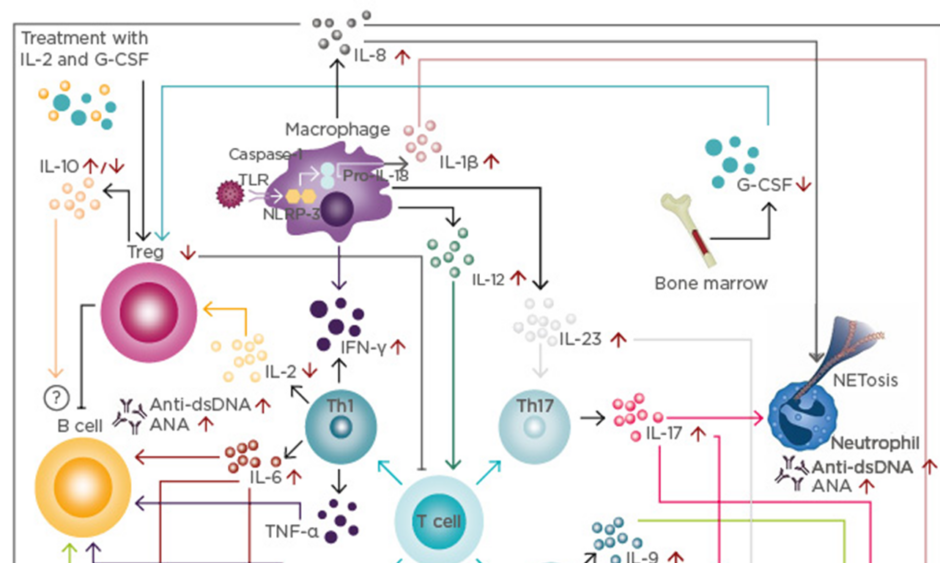
Figure 1: Cytokine network in systemic lupus erythematosus.
T cell response: Immune activation secondary to infections and other harmful agents triggers T cell responses inducing the differentiation of different T cell populations, such as Th1, Th2, Th9, and Th17.
B cell response: IL-4, IL-6, IL-9, and TNF-α produced by T cells increases the production of anti-dsDNA and ANA. Although IL-10 is considered a regulatory cytokine, its role on B cells is not fully understood.
Innate immune response: Activation of the inflammasome induces the production of IL-1β, which, together with IL-17, is associated with the induction of NETosis. Further, IFN-γ, IL-12, and IL-23 are associated with the enhancement of T cell responses, increasing the inflammatory process.
Modulatory immune response: IL-2 and G-CSF levels are decreased in patients with SLE, correlating with a reduction of Treg. Currently, clinical trials with IL-2 and G-CSF are underway.
SLE subphenotypes: Cytokines and innate and T cell responses influence the development of lupus nephritis, vasculitis, cutaneous lupus, arthritis, and neuropsychiatric SLE.
ANA: antinuclear antibodies; dsDNA: double-stranded DNA antibodies; G-CSF: granulocyte colony-stimulating factor; IFN: interferon; IL: interleukin; NETosis: neutrophil recruitment and extracellular trap formation; TLR: toll-like receptor; NLPR-3: NACHT, LRR, and PYD domains-containing protein 3; SLE: systemic lupus erythematosus; Th: T helper; TNF: tumour necrosis factor; Treg: T regulatory cells.
Cytokines and several inflammatory mediators are integrated in a complex network of biological elements, including genetic polymorphisms and environmental factors.9 Studies of the interactions between these variables from a systems medicine perspective could therefore provide a better understanding of the pathological mechanisms associated with the development of SLE. In this regard, Pacheco et al.10 found that some cytokine and autoantibody clusters (i.e., neutral, chemotactic/antiphospholipid antibodies, and Type 1 IFN-α/double-stranded DNA [dsDNA]) were associated with activity of disease. Thus, the understanding of the mechanisms associated with development of SLE is pivotal for the development of new interventions and accurate diagnosis strategies. Herein, the authors provide a summary of the most clinically relevant cytokines and inflammatory mediators associated with SLE.
CYTOKINES AND THE INNATE IMMUNE SYSTEM
Interleukin-1β
IL-1β is considered a multifunctional cytokine and belongs to the IL-1 family, a group of 11 cytokines, which plays a central role in the regulation of immune and inflammatory responses. IL-1β is characterised by highly inflammatory properties that may have a significant effect on disease.9 IL-1β is produced in response to inflammasome activation, which is commonly defined as a group of intracellular multiprotein signalling complexes associated with inflammatory responses to extracellular pathogens.11
It has been recognised that IL-1β levels are increased in patients with SLE, especially in individuals with systemic manifestations, such as fever.12 The activation of the inflammasome has been proposed as a mechanism in SLE pathophysiology, which includes the stimulation of toll-like receptors (TLR) and NF-κβ transcription by immune complexes or C3a.13
Polymorphisms of the IL1 gene have been associated with SLE development.14 IL-1β-deficient mice are resistant to induction of experimental SLE.15 Furthermore, IL-1β expression and IL-1 levels are increased in the kidneys of mice with lupus nephritis (LN).16 These data advocate for a plausible role of this cytokine in the development of LN, which may function as a diagnostic biomarker and therapeutic target in this subset of patients.17 Anakinra, an IL-1R antagonists, has been shown to be safe and well-tolerated in patients with severe lupus arthritis.18 However, the efficacy of this treatment is poor and the studies only include a low number of patients; thus, further studies are warranted to clarify the usefulness of this treatment in patients with SLE.
Interleukin-8
IL-8 is a potent neutrophil chemokine that has been previously associated with renal injury in humans.19 It has been found that IL-8 induces neutrophil recruitment and extracellular trap formation (NETosis), increasing the risk of antinuclear autoantibody production,20 suggesting it plays a role in the early stages of SLE.
IL8-845C polymorphism has been associated with an increased risk of LN in African Americans.19 Indeed, urine levels of IL-8 are associated with activity of SLE and LN.21 Furthermore, IL-8 concentrations in cerebrospinal fluid from patients with neuropsychiatric lupus were found to be higher than in healthy donors, and inflammatory mediators, such as IL-6, IP-10, and MCP-1, were simultaneously elevated, suggesting the existence of a complex interaction network among these inflammatory mediators and the development of neuropsychiatric lupus.22 Since long-term treatment of SLE with standard immunomodulatory drug regimens failed to normalise levels of key chemoattractant proteins linked to innate immunity, including IL-8,23 blockade of IL-8 could be considered a plausible therapeutic target in SLE.
Interleukin-12 and 23
The IL-12 family includes IL-12, IL-23, IL-27, and IL-35. These cytokines are critical for Th1 differentiation and play a major role as proinflammatory mediators.24 In a study conducted by Qiu et al.,24 patients with SLE showed high levels of these cytokines and were correlated with anti-dsDNA antibodies. The serum p40 monomer concentration of IL-12 was elevated in the sera of patients and correlated with the activity of disease.25 Clinical trials with IL-12p40 antagonist (ustekinumab) are underway.26
Regarding IL-23, patients with active disease showed higher IL-23 mRNA compared with patients with inactive disease, as well as healthy controls. Furthermore, IL-23 levels were significantly higher in patients with renal compromise.27,28 Further to this, mice treated with a neutralising anti-IL-23 antibody had less severe nephritis than control-treated mice, suggesting that IL-23 plays a role in the development of autoimmunity and ensuing inflammation in SLE.29 In vitro IL-23 treatment promotes IL-17 production and downregulates IL-2 production. The IL-23R knockdown mouse model presented fewer T follicular helper lymphocytes, B lymphocytes, and plasma cells, leading to decreased production of anti-dsDNA antibody.30,31 Similar to IL-12, clinical trials with IL-23 blockers (briakinumab) are ongoing.26
TYPE 1 T HELPER CELL RESPONSE AND SYSTEMIC LUPUS ERYTHEMATOSUS
Interleukin-2
IL-2 is an auto and paracrine growth factor for both T and B lymphocytes. This cytokine is considered an essential growth and survival factor for regulatory T lymphocytes (Treg).32 Nevertheless, it has been found that IL-2 downregulates the expansion of T follicular regulatory cells, thus suggesting the pleotropic role of this cytokine in immune responses and encouraging studies about the role of follicular IL-2 on SLE.32
It has been shown that lymphocytes from patients with SLE produce low levels of IL-2 that may trigger low counts of Tregs.33 This low production has been associated with active repression of IL-2 transcription mediated by the binding of phosphorylated CREM to the IL-2 promoter.32 In a study by Schorle et al.,34 IL-2/IL-2R knockdown mice showed SLE hallmarks, such as autoantibody production, lymphadenopathy, and decreased Treg lymphocytes. Recently, He et al.35 found that treatment with a low dose of recombinant human IL-2 selectively modulated the abundance of Treg lymphocytes, but not Th1 or Th2, and was accompanied by marked reductions in SLE disease activity.
Interleukin-6
IL-6 is a pleiotropic cytokine synthesised predominantly by monocytes, fibroblasts, endothelial cells, and less frequently in T and B lymphocytes. This cytokine induces the maturation of B lymphocytes and increases Ig secretion.36 The role of this cytokine in SLE has been widely studied and, in some cases, has been associated with SLE subphenotypes and disease activity.
It has been described that IL6-174G/C and IL6-572G/C polymorphisms are associated with the development of SLE.37 Talaat et al.38 found that patients with SLE showed higher levels of IL-6 than controls and that IL-6 was associated with activity of disease. Patients with lupus arthritis also showed high levels of IL-6, which, in turn, correlated with high anti-dsDNA antibody and ESR levels.39 A recent clinical trial reported by Illei et al.40 found that patients with SLE and arthritis who were treated with an IL-6 blocker (i.e., tocilizumab) exhibited an improvement of symptoms and some showed remission. Furthermore, patients treated with rituximab after B cell repopulation can be divided into two groups: those responding to rituximab (responders) and those not responding to rituximab (non-responders). Non-responding patients showed an increase in the expression of TNF-α and IL-6, and a reduction in CD24+ CD38high regulatory B cells compared to healthy controls and responders.41 All together, these data indicate IL-6 is a therapeutic target in SLE.
Tumour Necrosis Factor-α
TNF-α is a cytokine from a group of 15 proteins belonging to the TNF family. There are two active forms of the TNF-α protein, a membrane-bound form and a soluble form, and it is produced by a large group of immune cells, such as macrophages, T and B lymphocytes, natural killer cells, neutrophils, and astrocytes.42 TNF is considered a growth factor for B lymphocytes, inducing secretion of IL-1 and IL-6 and a triggering factor for the activation and proliferation of T lymphocytes.43
Controversial results in SLE have been found for TNF-α. Increased plasma levels of TNF-α were associated with immunomodulatory activity reducing severity but have also been linked to deleterious effects and increased disease activity.44-46 Indeed, TNF-α antagonists induce SLE-like disease, suggesting TNF-α is beneficial in a SLE context. Further studies are therefore warranted to clarify the role of TNF antagonists from a personalised medicine approach.
Interferon-α
IFN-α belongs to the Type I IFN family, which are glycoproteins known for their capacity to interfere in viral infections. The upregulation of the expression of these genes in SLE patients has been called the IFN-α signature.47 This cytokine has been associated with an increased number of plasma lymphocytes, autoantibody production, defective apoptotic cell clearance, and promotion of T cell-dependent inflammation.48 Three mechanisms of IFN-α response have been described. The first involves NETosis and the creation of an interferogenic signal mediated by TLR-7 or TLR-9. The second refers to the contribution of an extensive proportion of AD genetic risk variants that can affect IFN-α production.49 The third mechanism denotes a chronic dysregulation of plasmacytoid dendritic cells that induce an increase of IFN-α secretion.50,51 In murine models, the IFN-α pathway blockade is associated with better outcomes.52 Currently, promising anti-IFN-α monoclonal antibodies (i.e., sifalimumab and anifrolumab) are under clinical study.53
Interferon-γ
This cytokine is included in the Type II IFN group. Macrophages, natural killer cells, and T lymphocytes, especially CD4+ and CD8+ T lymphocytes, secrete IFN-γ. IFN-γ activates macrophages at the site of inflammation, contributes to cytotoxic T lymphocytes activity, has antiviral capacities, and has been strongly associated with Th1 response.47,54 Patients and murine models of SLE typically show high levels of this cytokine, and blockade abrogates SLE development in mice.9,55 An anti-IFN-γ monoclonal antibody is under development with promising results.56
TYPE 2 T HELPER CELL RESPONSE AND SYSTEMIC LUPUS ERYTHEMATOSUS
Interleukin-4
IL-4 is a pleiotropic cytokine characterised by the stimulation of CD4+ T lymphocytes to differentiate into Th2 and the inhibition of Th1-type cytokine production.57 It has been proposed that IL-4’s role in rescuing B cells from apoptosis may promote autoreactive B lymphocyte survival in mouse models.58 IL-4 treatment triggered the production of IgG anti-dsDNA antibody, and the blockade of IL-4 prevents the onset of LN.58 Furthermore, in SLE murine models, the IL-4 knockout mice produced less IgG1 and IgE serum Ig,59 thus suggesting a major role of this cytokine in the pathogenesis of disease.
However, evidence is conflicting, and some patients with cutaneous and articular manifestations showed low levels of IL-4,60 commonly due to an imbalance of IFN-γ/IL-4-producing CD4+ T lymphocytes.61 This ratio correlates with the activity of disease and is significantly higher among patients with LN.61
Interleukin-5
IL-5 is preferentially produced by Th2 lymphocytes. It is considered a growth and differentiation factor of eosinophils and B lymphocytes.62 Zhu et al.63 described that patients with LN and high activity of disease showed higher levels of IL-5 than controls. Furthermore, patients with severe or extensive skin lesions showed an overexpression of IL-5, suggesting that Th2 lymphocytes are involved in SLE skin inflammation.64 On the other hand, Timóteo et al.65 reported lower levels of IL-5 in patients with SLE than controls; however, these patients showed low activity of disease.
In murine models, the high expression of IL-5 may directly or indirectly mediate a skewed signalling of proliferation and differentiation of self-antigen-activated B lymphocytes, leading to suppression of AD.62 These data suggest that different concentrations of the cytokine could play a role in the development of SLE and the expression of diverse subphenotypes.
Interleukin-13
IL-13 is considered a strong anti-inflammatory cytokine, with modulatory properties that include the modulation of macrophages, monocytes, and B lymphocytes. Conversely, it has been found that patients with SLE showed high levels of this cytokine, especially in those with LN,66 thus suggesting a failure in immunomodulatory function of IL-13 in patients with SLE. In murine models, T lymphocytes infiltrating the glomeruli and perivascular areas predominantly produced IFN-γ, IL-13, and IL-17. Thus, IL-13 may also be an important factor in the pathogenesis of glomerulonephritis and vasculitis.67
Furthermore, increased CD38 expression in SLE T lymphocytes correlated with plasma levels of IL-13, and positively correlated with activity of disease, ESR, and serum levels of C3.68,69 DNA methylation levels within the IL10 and IL13 gene regulatory domains are reduced in SLE CD4+ T cells relative to healthy controls, and negatively correlate with IL10 and IL13 mRNA expression.70
TYPE 9 T HELPER CELL RESPONSE AND SYSTEMIC LUPUS ERYTHEMATOSUS
Interleukin-9
IL-9 is a T cell-derived factor preferentially expressed by CD4+ T lymphocytes with inflammatory properties. It has been found that patients with SLE showed high levels of IL-9 and the proportion of CD4+ IL-9-producing CD4+ T lymphocytes correlates with disease activity, proteinuria, low C3 titres, and high severity of disease.71,72 Similarly to IL-6, active patients who were treated and achieved disease control showed a reduction in IL-9 concentrations.73
Lupus-prone mice have shown an increased production of IL-9 and an expansion of Th9 lymphocytes, which were associated with anti-dsDNA antibody. In addition, IL-9 appears to promote B lymphocyte proliferation and autoantibody production, which could be blocked by inhibition of signal transducer STAT3. Indeed, IL-9 blockade reduced serum anti-dsDNA antibody titres and lessened renal disease.74
TYPE 17 T HELPER CELL RESPONSE AND SYSTEMIC LUPUS ERYTHEMATOSUS
Interleukin-17
IL-17 is a proinflammatory cytokine produced by activated T lymphocytes with a high influence to recruit monocytes and neutrophils.36 IL-17 can amplify the immune response by increasing the production of autoantibodies through the stimulation of B lymphocytes.75
Patients with SLE usually show high levels of IL-17A;76 in fact, the IL-17A serum levels positively correlate with activity of disease.77 Different subphenotypes have been associated with high levels of IL-17, including cutaneous, haematological, and central nervous system compromise.78,79 Furthermore, it has been shown that IL-17 is associated with LN and increased anti-dsDNA autoantibody production.80 The first report of an effective IL-17-targeted therapy in SLE was published recently.81
T REGULATORY CELLS AND SYSTEMIC LUPUS ERYTHEMATOSUS
Interleukin-10
IL-10 is a cytokine produced mainly by Treg cells and regulates the immune response; however, IL-10 improves B lymphocyte proliferation and Ig class switching, increasing antibody secretion.36 It has been described that patients with SLE showed high levels of IL-10, which were positively correlated with activity of disease and anti-dsDNA antibody, and negatively correlated with C3 and C4 levels, as well as with lymphocyte counts.82 In murine models, the IL-10 blockade limited the renal damage and decreased the production of anti-dsDNA antibodies.83
MRL-Fas(lpr) IL-10 knockout mice developed severe lupus, with earlier appearance of skin lesions, increased lymphadenopathy, more severe glomerulonephritis, and higher mortality than their IL-10-intact littermate controls.84 The increased severity of lupus in MRL-Fas(lpr) IL-10 knockout mice was closely associated with enhanced IFN-γ production by both CD4+ and CD8+ lymphocytes and increased serum concentration of IgG2a anti-dsDNA autoantibodies.84 Administration of the recombinant IL-10 reduced IgG2a anti-dsDNA autoantibody production in wild-type MRL-Fas(lpr) animals, supporting the protective effect of IL-10.84
In short, a pathogenic role for IL-10 appears to predominate and affect many facets of SLE, and its blockade is likely to prove an effective therapeutic strategy. However, no current clinical trials aiming to directly block IL-10 have been published.
OTHER INFLAMMATORY MEDIATIORS IN SYSTEMIC LUPUS ERYTHEMATOSUS
Erythrocyte Sedimentation Rate and C-Reactive Protein in Systemic Lupus Erythematosus
ESR and CRP are the oldest and most widely used biomarkers of systemic inflammation and tissue injury. Although CRP is replacing ESR as an inflammatory biomarker due to its high sensitivity and specificity, ESR remains a useful tool for diagnosis or monitoring of disease activity in SLE.8
ESR appears to be a reliable marker (with cut-off above 25–30 mm/h) for disease activity assessment in non-infected SLE patients. On the other hand, CRP >10 mg/L in noninfected patients could be indicative of disease severity. Additionally, in the absence of serositis or arthritis, a significantly increased CRP (>50–60 mg/L) is generally associated with infection.8
Osteopontin
Osteopontin is a pleiotropic protein that affects bone remodelling and immune system signalling. It plays a key role in regulating Th1/Th2 balance, stimulating B lymphocytes to produce antibodies, regulating macrophages and neutrophils, and inducing activation of dendritic cells. Overexpression of osteopontin has been associated with the pathogenesis of AD such as SLE.5 Recently, osteopontin-full and osteopontin N-half (cleaved from osteopontin-full) were measured in the serum and urine of SLE patients. In this study,85 osteopontin N-half in urine was higher in LN than in healthy controls. In addition, osteopontin N-half in urine decreased after the treatment of LN, suggesting that osteopontin N-half in urine could be a biomarker for evaluating activity of the disease. This data was confirmed in a meta-analysis, which revealed a significantly higher circulating osteopontin level in SLE patients, a trend of positive correlation between osteopontin levels and SLE activity, and a significant association between osteopontin gene 1239C>A and 9250C>T polymorphisms with SLE development.86
Soluble Urokinase-Type Plasminogen Activator Receptor
The soluble urokinase-type plasminogen activator receptor (suPAR) has been described as a valuable indicator of activity in the immune system. SuPAR is expressed in smooth muscle and endothelial cells.87 An inflammatory response leads to elevated suPAR levels, as reported in AD.88
Circulating suPAR has emerged as a potential marker of inflammation and disease severity in SLE. Recently, Enocsson et al.6 evaluated suPAR as a marker of disease activity and organ damage in SLE. The study found that suPAR levels were significantly elevated in patients with SLE compared to healthy controls. Furthermore, a strong association was detected between suPAR and severity of disease.6
Soluble Fas and Fas Ligand in Systemic Lupus Erythematosus
Fas and Fas ligand are members of the TNF and TNF-receptor families that are involved in apoptosis. The Fas receptor exists in two forms: one is attached to the plasma membrane, whereas the other is soluble (sFas). The latter is associated with anti-apoptotic functions.89 In SLE, sFas levels are increased due to a deletion in exon 6, and this increase has been associated with LN.7,90 Hatef et al.7 reported a significant rise in the serum concentrations of sFas and IL-18 in patients with proteinuria compared to those without it. Moreover, this study showed that the correlation between sFas and IL-18 in LN was significantly stronger than in mild SLE with similar non-renal SLE Disease Activity Index score.
CONCLUSION
Cytokines and inflammatory mediators are key factors for the development of SLE. As discussed in this review, cytokines are associated with diverse clinical manifestations (i.e., clinical subphenotypes), and some of them have been associated with activity and severity of disease. This suggests that cytokine profiles, if used as biomarkers, could aid in the monitoring and treatment of disease. However, it is necessary to recognise that these inflammatory mediators are associated with other biological variables that could reduce or increase their impact on different biological levels. Thus, additional studies to clarify those complex interactions are warranted. With the exception of belimumab, no Phase III trials of biologic therapy in SLE have met their primary endpoint. However, the significant steroid-sparing effect of these agents suggests that future trials may need to include steroid dose in a composite primary endpoint.91