Abstract
The sclerodermas are autoimmune rheumatic diseases associated with pathological fibrosis of tissues. The two forms, localised scleroderma (LS [also referred to as morphoea]) and systemic sclerosis (SSC), have different patterns of organ involvement depending upon age of onset. Juvenile LS (JLS) has a poorer prognosis than adult-onset LS (ALS), while juvenile systemic sclerosis (JSSC) has a better prognosis than adult-onset SSC (ASSC).
Optimal care requires appreciating the major differences between paediatric- and adult-onset disease, as they affect treatment and management strategies. Because the majority of patients with JLS have deeper tissue involvement, systemic immunomodulator rather than topical treatment is needed to mitigate their risk for serious morbidity and functional impairment. JSSC initially has a lower frequency of vital organ involvement than ASSC, but organ involvement can progressively accrue over time, so prolonged, aggressive treatment regimens may be needed. The authors recommend the care team for patients with JLS and JSSC include a rheumatologist who will be experienced in assessing and monitoring the most common extracutaneous involvement (musculoskeletal), as well as other organ involvement. Long-term monitoring of these patients into adulthood is essential; JSSC is a lifetime disease, while JLS can relapse or smoulder, with the disease activity focused in the deeper tissues.
The purpose of this review is to provide a clinically focused overview of JLS and JSSC disease patterns, highlighting differences between paediatric and adult-onset disease. The authors will review current care recommendations for JLS and JSSC, and discuss some of the challenges for their care, and areas for future research.
Key Points
1. Patterns of organ involvement for the two forms of scleroderma (localised and systemic) vary depending on age of onset. Juvenile localised scleroderma (JLS) has a poorer prognosis than adult-onset LS, while juvenile systemic sclerosis (JSSC) has a better prognosis than adult-onset SSC; JSSC is a lifetime disease, while JLS can relapse or smoulder.2. Paediatric- versus adult-onset disease therefore impacts treatment and management strategies, with a detailed understanding of the patterns of these diseases needed to direct optimal care.
3. Screening frequency for organ involvement, duration of treatment regimens, and long-term monitoring should consider paediatric onset for JLS and JSSC, rather than mirroring adult strategies.
INTRODUCTION
The sclerodermas are a family of autoimmune rheumatic diseases characterised by activation of the adaptive and innate immune system, genetic and vascular involvement, and dysregulated fibrosis.1,2 Both localised scleroderma (LS) and systemic sclerosis (SSC) are rare, with incidence of LS in the USA estimated to be 2.7 out of 100,000 persons,3 and the incidence of SSC worldwide estimated to be 1.4 out of 100,000 person–years.4 About one-quarter to one-third of all LS cases occur in children,3 whereas paediatric cases account for <5% of all SSC cases.5 As with most rheumatic diseases, there is a female predominance, but this is less pronounced for paediatric compared with adult-onset scleroderma (Tables 1 and 2).
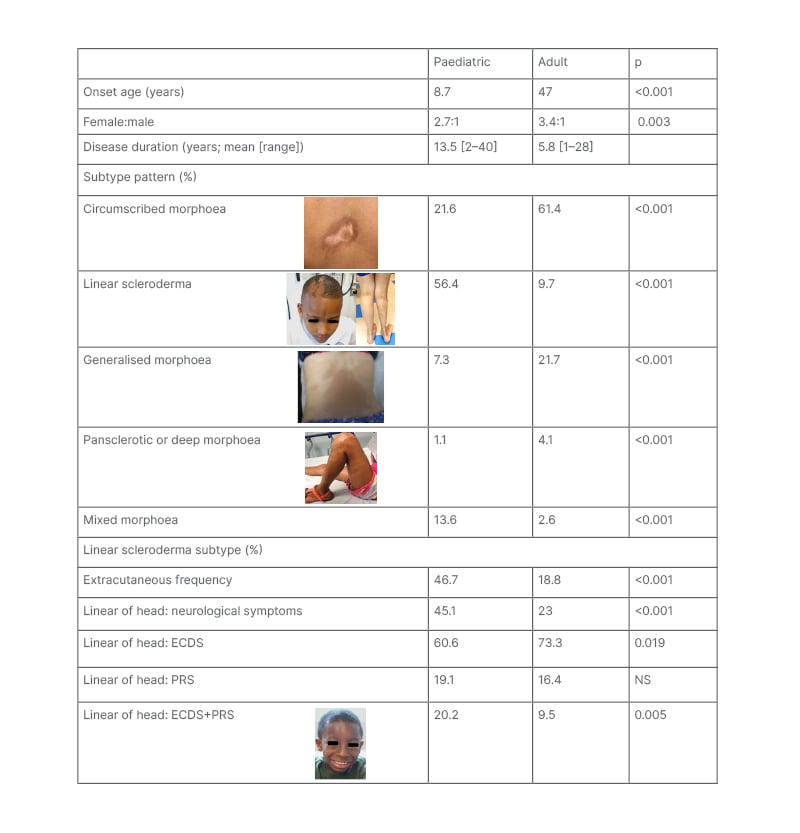
Table 1: Differences in subtype and extracutaneous patterns between paediatric- and adult-onset localised scleroderma.1-7
Subtype designation based upon the Padua preliminary classification criteria.6 Pansclerotic morphoea and deep morphoea were grouped together because several of the sources used for generating this table used a different classification criteria than the Padua Criteria.
ECDS: en coup de sabre; PRS: Parry–Romberg syndrome; NS: not specified.
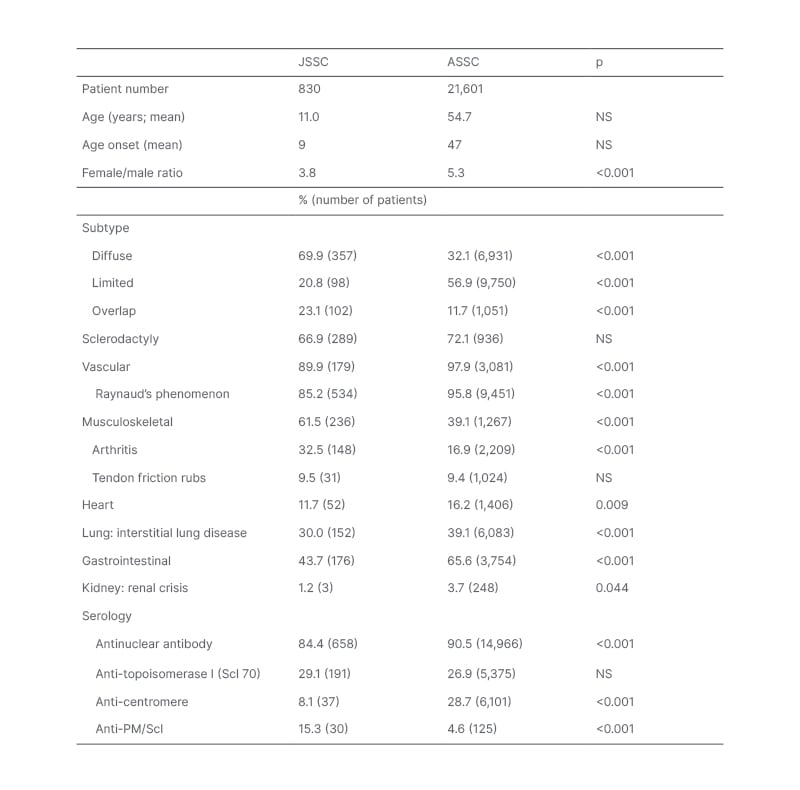
Table 2: Differences in subtype and organ involvement patterns between juvenile and adult-onset systemic sclerosis.
Data in the table was compiled from 19 JSSC studies5,8-25 and 10 adult SSC studies.26-35 The percentage affected was determined based upon cohort size for a given feature, with parenthesis indicating the reported number of patients affected. Studies differed in their terminology, so the authors scored the following as representing interstitial lung disease: pulmonary fibrosis, abnormal forced vital capacity, abnormal HRCT. P-values were calculated using the Z-score test for two population proportions (significant at p<0.05).
ASSC: adult-onset systemic sclerosis HRCT: high resolution CT; JSSC: juvenile systemic sclerosis; NS: not specified.
LS and SSC have distinct skin and organ involvement patterns with consequently different morbidity and mortality risks. Both diseases also have unique clinical patterns depending upon paediatric versus adult-onset. This review is focused on providing an overview of the current understanding of presentation patterns and morbidities associated with paediatric-onset LS and SSC. The authors’ review will compare the features of both groups, highlighting key differences in disease patterns, which are important to consider when deciding upon treatment and management. Paediatric rheumatologists treat most juvenile-onset patients with LS (JLS) with systemic immunomodulators to ensure adequate suppression of inflammation, and reduce the risk for damage development. This strategy has greatly improved outcome for JLS over the past decade, with a reduction in the frequency of arthropathy, limb length differences, and need for surgical intervention. On the other hand, understanding of how best to treat juvenile-onset SSC (JSSC) remains limited, due to the great rarity of this disease. The authors discuss some of the challenges for JLS and JSSC care, and areas for future research.
LOCALISED SCLERODERMA: CLINICAL FEATURES
LS, also known as morphoea, is recognised to have several subtypes that differ in skin lesion shape (ovoid, linear, or circumferential), lesion size, and disease extent (small or very limited to widespread). These subtypes are associated with differences in functional impact risks (nil to high). Most adults with LS have circumscribed morphoea, which is also known as plaque morphoea, and is the mildest subtype (Table 1). Plaque morphoea lesions are superficial, affect only the skin, and typically very limited in extent. The next most common adult LS subtype is generalised morphoea, consisting of larger plaque lesions that occur on at least two anatomic regions (head, anterior torso, posterior torso, right and left upper and lower extremities).36 Generalised morphoea lesions are usually also superficial in depth.6
The pattern is very different for paediatric-onset disease. Most patients with JLS have linear scleroderma, so-called because the lesions have a band-like appearance (Table 1).36 Linear scleroderma lesions usually affect deeper tissues such as muscle and bone. They can extend across the entire length of a limb, onto the torso, or across the face and scalp following an embryonic pattern known as Blaschko’s lines.37 Pansclerotic morphoea is the rarest and most severe LS subtype. Skin involvement is circumferential and confluent on the limbs, with extension often onto the torso and sometimes the head.36 Lesions often affect underlying tissues, predisposing the patient to chronic skin ulceration, with attendant risks of sepsis and squamous cell carcinoma.38 This subtype has been reported to be more common in JLS than adult-onset LS (ALS).36,38 Another subtype more common in JLS than ALS is mixed morphoea, which refers to a combination any of the other four subtypes (circumscribed, linear, generalised, or pansclerotic [hl]Table 1[/hl]). Most commonly, mixed morphoea presents as linear scleroderma with one of the other subtypes.1 Age-associated differences are also found within the linear scleroderma subtype, specifically for craniofacial linear scleroderma. Craniofacial linear scleroderma can present as a typical band-like lesion (en coup de sabre [ECDS]) or as progressive hemifacial atrophy. Progressive hemifacial atrophy, also known as Parry–Romberg Syndrome (PRS), affects deeper tissues without visible inflammation in the overlying skin.36 Compared to ALS, JLS has a lower frequency of ECDS but a higher frequency of the combination of ECDS and PRS (Table 1).
For most patients, severe morbidity is related to extracutaneous involvement, which is associated with functional impairment and higher physician damage scores.39,40 Extracutaneous involvement typically localises near the site of skin involvement, but presents remotely in 25–30% of patients.41 Onset of extracutaneous manifestations usually follow skin disease onset, with neurological involvement reported a mean of 4.3 years after.42 Late delays of 1–2 decades has also been reported,43,44 and about 16% of neurological problems precede skin disease.42
Functional impairment has been reported in 27–38% of patients with JLS. Most commonly, this manifests as musculoskeletal.39,45 Most patients with linear scleroderma of the limb or trunk have musculoskeletal impairment, from inflammatory (arthritis, myositis, fasciitis, tendonitis) and/or fibrosis related (joint contractures, angulation defects, muscle atrophy, limb length differences) problems.40,46 Patients with linear scleroderma of the head are especially at risk for neurologic, ocular, and oral morbidities, including seizures, peripheral neuropathy, uveitis, enophthalmos, and dental root defects.41,42,47
Extracutaneous manifestations are more commonly reported in JLS than ALS.47,48 In a retrospective study of patients with adult and paediatric LS, patients with JLS had a 32.5% frequency of musculoskeletal, ocular, oral, and neurologic morbidities compared with 8.0% in adults.7 Prospective studies have identified still higher frequencies of extracutaneous manifestations (46–74%) in JLS.40 This higher frequency in JLS than ALS partly reflects subtype differences, as linear scleroderma has a higher prevalence of deep tissue involvement than circumscribed or generalised morphoea (64% versus 32–46%, respectively).6
Age-associated differences in extracutaneous manifestations within the linear scleroderma subtype have also been identified. There was over a two-fold greater frequency of extracutaneous manifestations in paediatric compared to adult-onset linear scleroderma (47% versus 19%, respectively; p<0.001).1 Neurological involvement was also about twice as prevalent in patients with JLS versus patients with ALS craniofacial linear scleroderma, with higher frequencies identified for seizure, headache, and neuroimaging abnormalities.1 Many other severe neurological problems, including movement disorders, Rasmussen’s encephalitis, hemiplegic migraines, and cognitive and behavioural issues have been reported in JLS, but either very rarely or not at all, in ALS.42,48
The greater severity and higher frequency of extracutaneous manifestations in JLS compared with ALS is likely related to the disease spanning childhood, putting the child at risk for disturbed growth in affected areas during development. A two-fold higher frequency of extracutaneous manifestations in JLS was found for disease onset <10 years versus >10 years.49 Children with JLS can develop haemiatrophy of the affected body region (face, trunk, limb), joint contractures, and angulation defects. Furthermore, aberrant positioning of structures on affected sites such as the eye and teeth can lead to vision loss and malocclusion. Growth disturbances were identified in 39% of patients with JLS in a review of retrospective studies, and in 26% and 46% of patients, respectively, in two different prospective studies.40,50,51
JUVENILE LOCALISED SCLERODERMA TREATMENT
Paediatric rheumatologists are in consensus on systemic immunomodulator treatment for patients with active disease at risk for major morbidities.52,53 A recent Cochrane review supports methotrexate treatment for JLS, and this is also endorsed by the European Dermatology and Japanese Dermatology Associations.54-56 There has been one double blind, placebo controlled, randomised clinical trial of methotrexate treatment conducted in JLS, along with numerous case series and open label studies.51,57-59 Overall, the change from topical to systemic methotrexate treatment has been associated with major improvements in outcome. A comparison of patients with JLS pre-methotrexate to current cohorts showed a marked reduction in the frequency of joint involvement (50% to 20–23%, respectively), severely impaired function (22 to 11%, respectively), and orthopaedic surgical intervention (41% to 14%, respectively).50
Two paediatric rheumatology groups (Single Hub and Access Point for Paediatric Rheumatology in Europe [SHARE], and Childhood Arthritis and Rheumatology Research Alliance [CARRA]) generated standardised methotrexate regimens which are shown in Table 3.52,60 Three CARRA regimens were generated, which differ based upon corticosteroid inclusion and type; data was insufficient to support consensus on a single regimen. CARRA also generated criteria to define patients appropriate to treat with these regimens in treatment studies and tools to evaluate response, including for scoring skin activity and extracutaneous morbidity.51,52,61 Ideally, these regimens will be used in comparative effectiveness studies to identify the most effective regimen, and continue in an iterative fashion to identify the ‘best’ regimen.60 A pilot study, although underpowered for determining the relative effectiveness of the regimens, showed the feasibility of this approach, with all three regimens found effective.51
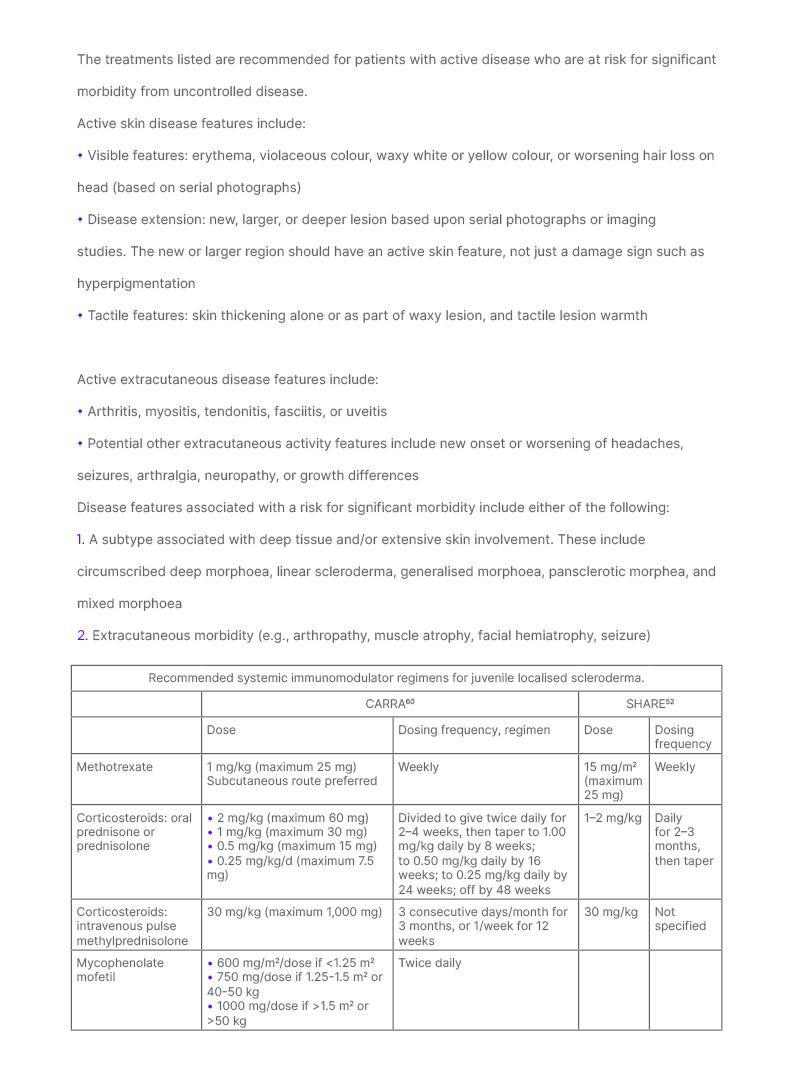
Table 3: Recommendations for treatment of patients with juvenile localised scleroderma at risk for significant morbidity.
Patients with JLS are more likely to develop major morbidity than those with adult onset LS due to the higher frequency of extracutaneous involvement and subtype pattern differences. Paediatric rheumatology organisations are in consensus to treat JLS patients with active disease at risk for significant morbidity with systemic immunomodulators. Criteria for active disease and patient characteristics associated with risk for significant morbidity were generated by the LS workgroup of CARRA for use in potential comparative effectiveness studies.60 These criteria were not intended to qualify or disqualify patients for any specific treatment.
Both CARRA and SHARE generated methotrexate-based treatment regimens for these JLS patients. CARRA generated three methotrexate dose regimens (consensus treatment plans [CTP]) that differ based upon inclusion and type of corticosteroid: methotrexate alone, methotrexate with oral corticosteroids, or methotrexate with intravenous corticosteroids.60 The three CTPs reflect best available evidence and current treatment practices of the CARRA membership. Current data is insufficient to support one CTP as superior, so CTP choice is the decision of the treating physician and family. SHARE has specified that methotrexate could be used with initial corticosteroid treatment, with general suggestions provided for corticosteroid dosing.52
For patients intolerant of or non-responsive to methotrexate, CARRA also generated a mycophenolate mofetil regimen that can similarly be used alone, or in conjunction with corticosteroids.60 Co-administration with methotrexate can also be done.
CARRA: Childhood Arthritis and Rheumatology Research Alliance; CTP: consensus treatment plans; JLS: juvenile localised scleroderma; LS: localised scleroderma; SHARE: Single Hub and Access Point for Paediatric Rheumatology in Europe.
Methotrexate treatment, with or without corticosteroids, is effective for almost 70% of patients.51,57 Factors associated with poorer response to methotrexate treatment include presence of extracutaneous manifestations, some subtypes (linear scleroderma, mixed morphoea, pansclerotic morphoea), and treatment delay.40,51,59,62 For patients who are non-responders, or intolerant to methotrexate, mycophenolic mofetil is most commonly substituted. Dosing regimens for mycophenolic mofetil were also generated by CARRA (Table 3.)60 Small case series have reported benefit for biologic agents such as abatacept and tocilizumab for JLS (reviewed in Vasquez-Canizares N et al.;1 a more detailed discussion of treatment management, including an algorithm, can be found here).
Duration of treatment is commonly 2 or 3 years, but relapses still occur in 22–44% of patients.62-64 Re-treatment is effective at controlling relapse, but some patients will have persistently active, chronic remitting/relapsing, or evolving disease for decades.45,65 Despite improvements in treatment strategies, >25% patients with JLS still have functional impairment, bone size difference, and/or joint limitation.40,51
SYSTEMIC SCLEROSIS: CLINICAL FEATURES
There are several differences between JSSC and adult-onset SSC (ASSC), including gender (lower female predominance in JSSC), subtype predominance, organ involvement, and autoantibody profile. Table 2 presents data compiled from 19 JSSC studies,5,8-25 and 10 adult SSC studies.26-35 The JSSC studies were selected based upon a limited literature review, and include the largest recent cohorts that described subtype and organ involvement. Several older international cohorts that reported on a minimum of three patients with JSSC were included. The adult studies were selected based upon their inclusion of large number of patients where the frequency of most organ systems was described, selecting cohorts representing different international populations.
Limited cutaneous is the most common ASSC subtype, followed by diffuse cutaneous. In contrast, diffuse cutaneous is the most common paediatric subtype, followed by overlap (Table 2). As expected from the lower frequency of limited cutaneous subtype, there is a much lower frequency of anti-centromere antibody in JSSC than ASSC. No age-related difference was found for the frequency of anti-Scl70 positivity (Table 2). A higher frequency of anti-PM/Scl antibody positivity was identified in JSSC, which likely partly reflects the greater frequency of the overlap subtype.
The frequency of sclerodactyly is similar across ages, while other musculoskeletal involvement is more common in JSSC (Table 2). Patients with JSSC have a lower frequency of vital organ involvement than ASSC, resulting in a lower mortality rate (10 years mortality rate: 15% for JSSC, 34% for ASSC in 2002).14,66 The most common mortality patterns in JSSC is rapid disease progression that results in death within 5 years of diagnosis.67
Gastrointestinal involvement in JSSC is common, with low BMI and weight loss frequently reported.10,16-19 Compared with other paediatric rheumatology diseases, JSSC was associated with the lowest body mass index Z scores, with 28% of patients with JSSC having a Z score of -1 or lower.19 As with adults, oesophageal involvement can be asymptomatic, or associated with dysphagia, gastroesophageal reflux disease, and retrosternal pain.68 Also, similar to ASSC studies, oesophageal involvement in JSSC is associated with lung involvement, such as lower forced vital capacity, and pulmonary symptoms (dyspnoea, cough).68
JUVENILE SYSTEMIC SCLEROSIS TREATMENT
Consensus recommendations for JSSC care were recently published by a SHARE group.69 The rarity of JSSC has made treatment studies difficult, so recommendations are generally based upon descriptive case-control studies, or expert opinion.69 Lung evaluation using high resolution computerised tomography and pulmonary function test with diffusing capacity for carbon monoxide is recommended, and routine pulmonary function test monitoring is also recommended at least every 6 months. Cardiac (echocardiogram), skin, and renal monitoring should be monitored at a similar frequency. For JSSC, there is a need for reliable and validated outcome measures. An international effort is currently underway to develop consensus outcome measures based upon a systematic literature review, surveys, and Delphi process. This effort should help to standardise care, and enable international comparative studies.70
The SHARE panel recommends that treatment with systemic immunomodulatory drug(s) be considered for all patients with JSSC at diagnosis.69 They recommend systemic corticosteroid treatment be considered in addition to a disease modifying anti-rheumatic drug. The high frequency of arthritis and rarity of renal crisis in patients with JSSC (Table 2) supports the use of systemic immunomodulators, including corticosteroids, for these patients. The rise in prevalence of pulmonary fibrosis in patients with JSSC over time, rising to 63% in diffuse cutaneous and 14% in limited cutaneous at 20 years, also supports this treatment strategy.71 Case studies of refractory patients with JSSC have reported impressive benefits for lung and heart disease from tocilizumab or rituximab treatment.72,73 The tocilizumab treated patients previously failed cyclophosphamide and mycophenolate mofetil treatment, and were still able to respond well to tocilizumab a mean of 6.9 years later.72 Nintedanib, an anti-fibrotic agent found effective for slowing lung progression in adult patients with SSC, was recently approved for treatment of adult SSC interstitial lung disease.74 It is currently being studied in a double blind, placebo controlled, randomised clinical trial in paediatric patients with interstitial lung disease,75 and may be available for treating patients with JSSC in the near future.
CONCLUSIONS
Paediatric-onset localised scleroderma and systemic sclerosis both differ from adult-onset disease in several major clinical features. These major differences imply the need for paediatric scleroderma specific care and treatment strategies, rather than relying solely upon adult strategies. It is important for adult providers to appreciate these differences so they can provide the appropriate care and monitoring for these patients when they transition to adult care. Because musculoskeletal involvement is present in the majority of patients with JLS and JSSC, the authors recommend that care teams for these patients include a rheumatologist, who will be able to identify the development and progression of musculoskeletal and other extracutaneous involvement. Other subspecialists are also often needed for care, especially for patients with paediatric-onset SSC.
JLS is more severe than adult-onset LS, with a higher prevalence of extracutaneous involvement and longer active disease duration that spans childhood. These features put patients with JLS at risk for functional impairment and disfigurement from a wide range of morbidities, such as limb length differences, arthropathy, seizures, and facial hemiatrophy. Treatment to control active disease with methotrexate and other systemic immunomodulators is currently the best strategy to limit the risk for, and level of damage, and has greatly improved outcomes. Relapses are common, and may even present remotely as new or worsening extracutaneous morbidity, so long-term monitoring of these patients through adulthood is vital.
JSSC has a lower mortality rate than adult-onset SSC, but still ranks as one of the most severe paediatric diseases due to substantial morbidity from skin, musculoskeletal, gastrointestinal, and lung disease. JSSC may have a higher incidence of inflammation than adult-onset disease, with a higher frequency of overlap subtype and arthritis. Lung disease may also continue to progress over decades in JSSC patients, so long-term aggressive treatment may be warranted to minimise morbidity and mortality risks. Recent consensus care recommendations specify that all new patients should be considered for systemic immunomodulator and corticosteroid treatment.
Overall, more research is needed for both JLS and JSSC. Paediatric rheumatology organisations have generated several treatment regimens and measures for JLS to assess response. More JLS treatment studies, including comparative effectiveness studies, are needed to identify the most effective regimens, especially for patients that continue to have functional impairment, growth disturbances, neurological symptoms, or who are methotrexate non-responders, or are methotrexate intolerant. Research in JSSC has lagged behind JLS due to the greater rarity of JSSC, and the lack of consensus outcome measures. A current international effort to generate consensus measures that can be used in international JSSC treatment studies should advance care by enabling more data-based recommendations. The enormous, continued advances in adult SSC knowledge of disease pathophysiology and treatment also offer great hope and promise to improve the outcome for patients with paediatric-onset disease.